Double success for huntingtin RNAi gene silencing
2 bits of good news for RNAi gene silencing in HD: it's safe over six months, and a way to treat bigger brain areas
 By Professor Ed Wild January 24, 2012 Edited by Dr Jeff Carroll
By Professor Ed Wild January 24, 2012 Edited by Dr Jeff Carroll
Most HD researchers are pretty excited by the idea of ‘silencing’ the Huntington’s disease gene, to reduce production of the harmful huntingtin protein. Two challenges - safety and delivery - are now closer to being solved thanks to collaborative work by academic and industry researchers.
We’re big fans of gene silencing. Like many Huntington’s disease researchers, we think it’s the approach most likely to produce an effective treatment for HD.
Gene silencing involves using a specially designed drug to intercept a message molecule, called RNA, that’s produced from the HD gene and tells cells to make the harmful huntingtin protein. The effect of the drug is that cells make less of the protein.
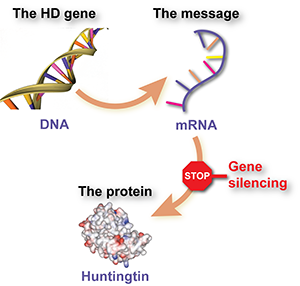
Put even simpler, gene silencing is like a stop sign for mutant huntingtin.
Rapid progress
So far, gene silencing for HD, in various forms, has cleared every hurdle it’s encountered. It’s now been tested in several mouse and rat models of HD, and not only slowed down progression but actually produced improvement in both symptoms and brain damage. It seems that the brain can actually recover - to some extent - if only levels of the harmful protein can be lowered a bit.
The remaining hurdles
Several research teams are heading for human trials of gene silencing in HD. But there are a few remaining challenges to address before that can happen.
The first is safety. Unexpected side effects are always possible, and could be dramatic, since we’re talking about drugs that are injected or infused directly into the nervous system, and interact directly with our cells’ genetic machinery.
Another is delivery. Gene silencing drugs can’t be given as pills or injections into the blood, because they wouldn’t be let into the brain. So they have to be introduced directly into the nervous system. Depending on the structure of the drug, this means an operation to introduce needles or tubes into either the spine or the skull. Sounds drastic, but if the treatment is effective, it’ll be worth it.
The delivery problem doesn’t stop there though, because once the drug is in the head, it has to get inside our brain cells to work its genetic wizardry.
The gene silencing menu
Gene silencing researchers have to make several choices before they begin a treatment trial. Here’s a checklist to help you understand future news stories.
First, we have to decide on the structure of the drug. The two basic choices are RNAi drugs, which are chemically similar to the body’s RNA message molecules; and ASO drugs, which are slightly different but may be better absorbed by cells.
The second choice is what target to choose: both copies of the HD gene, or just the mutant one. Targeting both - called non-specific silencing - is easier, but switching off the ‘normal’ gene might be dangerous. Targeting just the mutant gene - called allele-specific silencing - may be safer but is much harder to do.
The third choice is destination - where will the drug go? RNAi drugs don’t naturally spread far, so the drug has to be delivered right into the substance of the brain. Drugs that spread further, like ASOs, could be infused into the fluid around the brain or, if we’re lucky, at the base of the spine.
Fourth, we need to decide on delivery. Will the drug be given on its own, packaged in a virus or pumped at pressure, to increase its spread through the brain?
The fifth choice is treatment regime. Should we give a one-off treatment or infuse the drug over weeks or months? Right now we don’t know how long the effects will last, so this has to be worked out by comparing different regimes.
Whenever you read about gene silencing research, it’s helpful to figure out up front, which option has been selected for each of these choices.
Three come along all at once
At the recent HD World Congress in Melbourne, HDBuzz reported on exciting presentations from several gene silencing research groups. Then in November, we brought you news of the first safety trial of huntingtin gene silencing using RNAi in a primate brain.
“The RNAi drug spread further than you’d get with simple injections. A lot further, in fact. ”
Now, two further scientific papers have been published - each the fruit of collaboration between academic researchers and biotechnology companies. Both papers involved Minneapolis-based company Medtronic and the team of Dr Zheming Zhang at the University of Kentucky.
Six-month safety
The November report of RNAi safety in primates was quite a short study - six weeks. The new study by Medtronic and Zhang’s team, reported in the journal Brain, was also done in rhesus monkeys, but lasted a full six months.
Let’s look at what the researchers studied, using the checklist above:
Structure: this was a trial of an RNAi drug.
Target: both copies of the gene were targeted - non-specific knockdown.
Destination: the substance of the brain - the striatum, to be exact, which is affected early in HD patients.
Delivery: the drug was packaged into an empty virus, called AAV2.
Treatment regime: a one-off injection into five sites on each side of the brain
It’s also worth noting that ‘normal’ monkeys were used, with no expanded copies of the HD gene. So, this trial could only measure protein changes and safety - it can’t predict improvement in patients.
After treatment, the monkeys were observed for six months, looking at their general health and movement control. The surgery was well tolerated, and no new problems were seen in the treated animals.
As hoped, levels of huntingtin protein fell significantly in the treated regions. At each injection site, protein levels were reduced over an area about six millimeters across - in volume, that’s about three M&M’s worth per brain. It may not sound like much, but in a human brain that could make a big difference, and remember these measurements were made 6 months after the one-off treatment.
Thankfully, the drug caused no harmful brain changes like inflammation, infection or neuron damage.
So, the drug did its job of reducing protein levels, and treatment didn’t appear to produce any harmful effects. The authors reckon that six monkey months equates to about 18 human months. Sounds good - but as the authors point out, it could take even longer in humans for good or bad effects to emerge.
An innovative delivery method
The other new paper came from a three-way collaboration between the University of Kentucky team, Medtronic, and RNAi drug company Alnylam Pharmaceuticals. It was published in the journal Experimental Neurology.
This was also a study of non-specific knockdown RNAi, delivered into the striatum of monkeys.
What distinguishes this work is the innovative delivery method. A technique called convection enhanced delivery (CED) was used. This involves placing tubes through the skull and into the substance of the brain. The top end of the tube is connected to a small pump that constantly squirts the drug down the tube under pressure. This pressure is the key - it causes the drug molecule to spread much further than it otherwise would.

CED is already used to get chemotherapy drugs to spread further within brain tumors. But would it work to deliver an RNAi drug?
First, the drug was pumped into the brain for seven days. Only one side of the brain was treated, so that the other could be used for comparison. A range of doses and infusion rates was used, to find the best combination. Then, a 28-day infusion was tried. Cleverly, the team made harmless radioactive modifications to the drug, that enabled them to measure exactly how far it had spread.
The drug did its job of reducing huntingtin levels, and the tubes and infusions didn’t particularly harm the brain.
But was the additional hassle of fitting tubes and pumps worth it - did the drug spread further? In short, yes.
Measurements showed that the RNAi drug reached much further than you’d expect with simple injections. If both sides of the brain had been treated, huntingtin levels would have been reduced in about eleven M&Ms’-worth of brain.
In a nutshell…
Before these two papers came out, we already knew that huntingtin could be lowered in the monkey brain using RNAi. Now we can add two major check-marks to our wish-list: first, silencing works and is safe over longer periods, and second, there are ways of getting the drug to spread further.
What could go wrong?
Cautious optimism is a wise approach here. There are certainly some things that could go wrong on the way to human trials, or during them.
These primate trials have shown that lowering huntingtin levels is safe in healthy monkeys. But that doesn’t mean it’s definitely safe in human patients. Human brains are much bigger and more complex than monkey brains. So the treatment could be less effective, or more dangerous, simply because of the species difference.
It’s also possible that the healthy protein somehow protects the brain from its harmful brother in humans. If that’s the case, silencing both copies could unexpectedly do more harm than good.
The brains of people with HD symptoms are probably more fragile and difficult to operate on, too. The bits of the brain that need treating are smaller than normal, because of shrinkage caused by HD. So the operations may be harder and more risky.
Finally, detecting success might be difficult in humans, because the disease progresses slowly, and we can’t examine patient brains under the microscope.
But never forget - all of these problems are being worked on together by some of the best scientific minds in the world, all focused on making effective treatments a reality for patients.
2012 - the year of gene silencing for HD?
Could 2012 be the year of gene silencing for HD patients? Will we see one or more human trials in the coming months? On the strength of progress so far, we actually believe that’s a reasonable thing to hope for, and several groups are working hard to make it a reality. The first trials will be small, and will be carried out slowly and with great caution, because safety is the prime concern. But if all goes well, larger trials will follow.


