
Huntington’s disease therapeutics conference 2022 – Day 1
Check out research updates from Day 1 of the 2022 HD Therapeutics Conference #HDTC2022



Good morning from sunny Palm Springs! After a 2-year hiatus because of COVID, the HD Therapeuticstherapeutics treatments Conference is back in person this year – the biggest annual gathering of HD researchers! Our Twitter updates are compiled below. Continue to follow live updates for the rest of the conference with the hashtag #HDTC2022.
Day 1 is focused on research updates from some of the top HD labs around the world.
Huntingtin proteinhuntingtin protein The protein produced by the HD gene. building blocks
Dr. Paolo Beuzer (CHDI) and Dr. Vanessa Wheeler (MGH) are introducing the first session of research talks, which will focus on ways to study and potentially manipulate CAG repeats in huntingtin DNA and RNARNA the chemical, similar to DNA, that makes up the ‘message’ molecules that cells use as working copies of genes, when manufacturing proteins..
CAG repeats – more complex than they seem
The first speaker of the day is Darren Monckton from the University of Glasgow. The Monckton lab researches the repeats in the DNA sequence in diseases like Huntington’s disease.
While the CAG repeatCAG repeat The stretch of DNA at the beginning of the HD gene, which contains the sequence CAG repeated many times, and is abnormally long in people who will develop HD seems simple because it’s so short, it’s actually quite complicated. The size of the CAG repeatCAG repeat The stretch of DNA at the beginning of the HD gene, which contains the sequence CAG repeated many times, and is abnormally long in people who will develop HD alone doesn’t account for the age that someone will develop HD symptoms. CAG codes for the protein building block glutamineglutamine the amino acid building block that is repeated too many times at the beginning of the mutant huntingtin protein. But other protein letters can also code for glutamineglutamine the amino acid building block that is repeated too many times at the beginning of the mutant huntingtin protein. One of those is CAA, which can also contribute to the polyglutamines in HD. These CAA interruptions can also affect the age at which someone gets symptoms. Having a CAA interruption, which is rare, causes an earlier age of onset compared to HD individuals who only have pure CAG repeats. Changes in the “purity” of the CAG repeatCAG repeat The stretch of DNA at the beginning of the HD gene, which contains the sequence CAG repeated many times, and is abnormally long in people who will develop HD tract i.e. whether it has these interruptions or not, could affect a process called somatic instabilitysomatic expansion A process in which the CAG repeat in the Huntingtin gene can change over a person’s lifetime in some cells of the body, particularly in the brain. which we wrote about here: https://en.hdbuzz.net/291
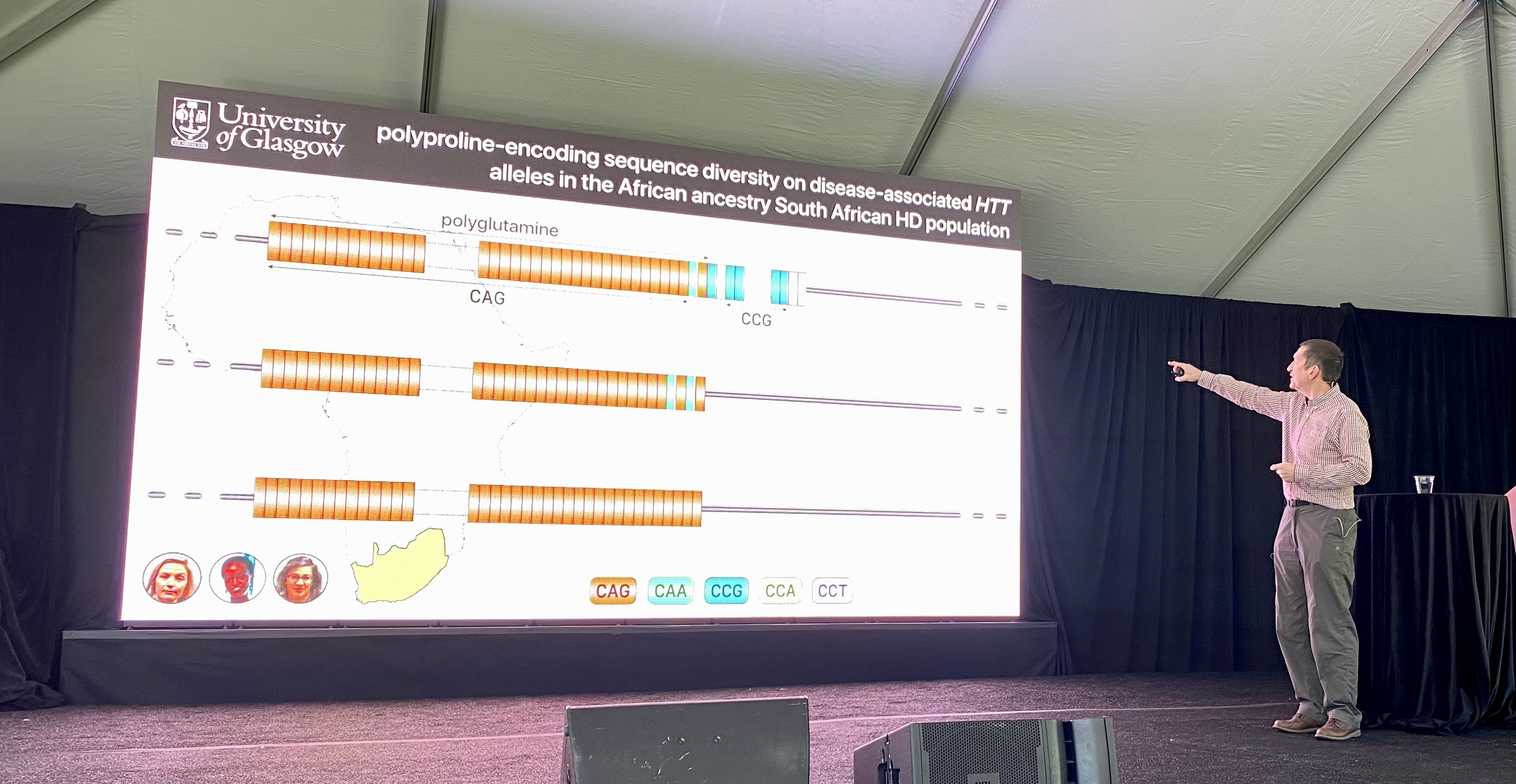
The EnrollHD database from which these observations are made, is biased with data from lots of European and North American people, but not from other parts of the world. The Monckton lab decided to tackle this by teaming up with a group in South Africa. In this South African population, the Monckton lab saw a very similar distribution of the different types of CAG tracts, with or without these interruptions. However there were some differences in another part of the HTTHTT one abbreviation for the gene that causes Huntington’s disease. The same gene is also called HD and IT-15 gene…
After the CAGs, the HTTHTT one abbreviation for the gene that causes Huntington’s disease. The same gene is also called HD and IT-15 protein contains letters, CCG, that make up a protein building block called proline. The Monckton lab sequenced how many prolines HD patients in South Africa had and found this area of the protein differs from HD patients with European descent. They used this data to look at how the number of proline repeats and the letters that make up those proline repeats affect age of onset. People with HD whose prolines who had a slightly different spelling had a 10 year earlier onset of HD symptoms. Tracking the way these protein building blocks are spelled can help improve diagnosis or prediction of age at disease onset. Overall, what this means is that other changes in the huntingtin genetic recipe can affect the disease – HD genetics is proving to be much more pesky and complex than it first seems. Understanding these variations which lead to earlier or later symptom onset might help researchers find new ways to make medicines for people with HD – that’s the hope.
The researchers conclude that these other changes in the huntingtin recipe are not affecting somatic instabilitysomatic expansion A process in which the CAG repeat in the Huntingtin gene can change over a person’s lifetime in some cells of the body, particularly in the brain. but perhaps are affecting the message made from the huntingtin recipe. The mRNAmessenger RNA A message molecule, based on DNA, used by cells as the final set of instructions for making a protein. message molecule structure might be changing. Interestingly, none of the changes we’ve just described change the huntingtin proteinhuntingtin protein The protein produced by the HD gene. itself. They only change the spelling of the gene or “recipe”. This suggests that it’s not protein-related changes that are affecting disease, but rather changes at the RNARNA the chemical, similar to DNA, that makes up the ‘message’ molecules that cells use as working copies of genes, when manufacturing proteins.-level. Altering the spelling of the RNARNA the chemical, similar to DNA, that makes up the ‘message’ molecules that cells use as working copies of genes, when manufacturing proteins. can change the way the molecule folds. No one yet knows what those folding changes mean, but they could be leveraged to develop therapeuticstherapeutics treatments.
Dr. Monckton concludes, even though it all seems so simple – that people with HD have increased CAG repeats – it’s actually very complicated! But research like this gets at how we can take advantage of this complexity to design new drugs.
Examining how HD affects individual cells in the brain
Next up is Steve McCarroll who is affiliated with the Harvard Medical School and Broad Institute. Steve will be telling us about his research on understanding HD at the level of single cells in the brain.
The brain is made up of lots of different cell types that perform specific functions. Dr. McCarroll highlights the need to understand how these many different types of cells are affected by HD. His lab uses specialized techniques to separate out different types of cells and understand their genetics. They are committed to sharing the methodology widely to benefit the entire HD research community.
Dr McCarroll has found a way to speed up his analysis – he combines human brain samples from HD patients together, then separates the data out after. Getting data faster is a big advantage because it allows researchers to get their answers as fast as possible. These types of large scale analyses are made possible through brain donations after a person with HD passes. Brain donations to HD research are a major way the field can get answers about HD in the only organism we care about curing HD in – people.
The McCarroll lab is applying these techniques to understand how the proportion of different kinds of cells in the brain changes as HD symptoms progress. His lab has defined these changes as the disease progresses, which helps us understand the cellular composition of the brain in people with HD. In most people with HD, there is significant loss of cells called medium spiny neuronsneuron Brain cells that store and transmit information. Researchers have known this for a while, but Dr McCarroll has also shown there are cellular changes in many other cell types in the brain. The loss of cells is accompanied by changes in which genes turn on and off. Dr. McCarroll has mapped these changes in these genes in each cell type as the disease progresses – wow!
These types of data can identify different genes within specific cells that modify the disease. One of those disease-related modifications is associated with expansion of the CAG repeatCAG repeat The stretch of DNA at the beginning of the HD gene, which contains the sequence CAG repeated many times, and is abnormally long in people who will develop HD as a person with HD ages.
Certain people with HD have an increase in their CAG repeats over time, particularly in the brain. These expansions can increase the age of onset for HD patients. Understanding what causes these expansions could help develop medicines to delay disease onset. Other genes, known as genetic modifiers, affect whether and how much a person’s CAG repeats will expand over time. McCarroll’s lab is looking at these modifiers within individual cells in many people!
Knowing how these processes change at such a small level produces a LOT of data that will give tons of information about how CAG expansion is changing in various cell types and how that affects disease progression. Interestingly, he found these CAG expansions happen to a much greater extent in medium spiny neuronsneuron Brain cells that store and transmit information, which are one of the most affected cell types by HD. This could be one of the reasons why this particular cell type is so vulnerable in HD. He’s also defined these changes in different cell types of the brain as well as different areas of the brain. Depending where in the brain a certain cell type is can also affect CAG expansion in that cell type. So it’s not just cell identity, but also cell location that matters! Understanding why both cell type and “neighborhood” in the brain affect CAG expansion will be an important next step towards developing therapies to combat it. This data from the McCarroll lab is hot off the presses and reflects recent breakthroughs in laboratory and analysis techniques. They plan to apply these techniques to more samples from people with various stages of HD.
HD mouse models
The next speaker is Dr. William Yang from the University of California, Los Angeles who will be telling us about his new mouse model, which his lab recently developed. We recently wrote about this new model: https://en.hdbuzz.net/318
No HD mouse model is perfect for studying HD, but different types can capture different aspects of the disease and allow for different types of experiments. For many years the Yang lab has specialized in creating mouse models to answer specific questions about HD. Choosing the right model for specific experiments is critical, since some mouse models only have certain features of HD – like altered gene expression or protein aggregation. The main innovation of the
Yang lab’s new mouse model is that it shows somatic instabilitysomatic expansion A process in which the CAG repeat in the Huntingtin gene can change over a person’s lifetime in some cells of the body, particularly in the brain., the growth of CAG repeats in certain cells over time. This allows researchers to understand the consequences of expansion to the health and behavior of the mice.
In these mice, the more CAG repeats expand, the more their behavior and brain cell health are affected, confirming for the first time in animals what we have suspected based on data from human blood, spinal fluid, and brain donations.
The lab is now using their new mouse model to better understand how unstable, expanding CAG repeats affect the huntingtin recipe and protein and the harm they may be doing in cells.
Dr. Yang also shared data from a different type of HD mouse model which is allowing them to study how genes get turned on and off over the course of HD. It’s great to see this question approached from multiple angles (along with the McCarroll lab and others). We’re taking a quick break now but will be back shortly with updates from the rest of this morning’s speakers. Stay tuned!
Processing the huntingtin message
Our next speaker is Dr. Gillian Bates from Queen Square Institute of Neurology, University College London. Dr. Bates will be updating us on how the huntingtin gene is processed and how we can perhaps use this information to develop therapeuticstherapeutics treatments.
The huntingtin gene gets “spliced” to remove small bits of genetic information that sit between the code. The gene then gets put back together before the protein is made. This process is typically used to give cells diversity in the information they can create from a single gene. But this process can go wrong in HD. In HD, the huntingtin gene is used to create a very small fragment of a protein – called “exonExons The small fraction of our DNA that is directly used to instruct cells how to make proteins 1”. This exonExons The small fraction of our DNA that is directly used to instruct cells how to make proteins 1 contains the CAG repeats and is very toxic to cells.
Dr. Bates looked at the amounts of exonExons The small fraction of our DNA that is directly used to instruct cells how to make proteins 1 in HD mouse models and in different areas of brains from people with HD. She found that with longer CAG repeats, the splicingsplicing the cutting up of RNA messages, to remove non-coding regions and join together coding regions. process making the exon1 protein happened more frequently. The Bates lab is experimenting with ways to detect and distinguish between different forms and pieces of the huntingtin proteinhuntingtin protein The protein produced by the HD gene. created by splicingsplicing the cutting up of RNA messages, to remove non-coding regions and join together coding regions.. They do this using different combinations of antibodies, a way to detect different parts of the protein. This work suggests the exonExons The small fraction of our DNA that is directly used to instruct cells how to make proteins 1 fragment is the site of protein aggregation creation. Understanding how this process occurs can give us lots of clues about how to reduce these protein clumps.
The Bates lab specializes in innovative ways to try and see different forms of the protein under a microscope or in an assay, like creating novel mice and treating the tissue with different chemicals. They made a special mouse model where the splicingsplicing the cutting up of RNA messages, to remove non-coding regions and join together coding regions. pattern is altered, and the exon1 fragment of the huntingtin proteinhuntingtin protein The protein produced by the HD gene. should no longer be made. In these mice the lab looked at the levels of toxic protein clumps which are made compared to regular HD mouse models. In the new mouse model there were a lot less clumps suggesting the exon1 fragment is important for making the clumps. Next steps will involve exploring how differences in huntingtin clumping could change mouse behavior and the pattern of communication between brain cells. Sometimes huntingtin clumps show up near the cell’s nucleusnucleus A part of the cell containing genes (DNA) – the part of the cell that houses genetic material. The Bates lab used multiple models to show that this only happens with human huntingtin, not mouse huntingtin. These data suggest there’s something unique about human huntingtin that leads to these pathogenic protein clumps. This may be a clue to why humans are the only species to naturally get HD!
Understanding which forms of huntingtin are most toxic and why will help us design drugs to combat its negative effects in (human!) brain cells.
Cellular handling of the huntingtin proteinhuntingtin protein The protein produced by the HD gene.
Next up is Dr. Judith Frydman from Stanford University. She’ll be talking about why CAG repeatCAG repeat The stretch of DNA at the beginning of the HD gene, which contains the sequence CAG repeated many times, and is abnormally long in people who will develop HD expansions can lead to problems with “trash tagging and disposal” systems in brain cells.
While we know the cause of HD, researchers don’t truly know the “normal” function of the huntingtin proteinhuntingtin protein The protein produced by the HD gene.. What they do know is that it participates in a variety of different biological processes – kind of like a swiss army knife of the cell. Because of this, researchers debate if HD is a disease caused by disruption of other genes or a disruption of other proteins. Dr. Frydman’s work argues that HD, at least in part, results from disruption at the protein level.
Dr Frydman’s research focuses on understanding how the huntingtin message molecule, called mRNAmessenger RNA A message molecule, based on DNA, used by cells as the final set of instructions for making a protein., is turned into the protein molecule, through a process called translation. Stresses on cells (things like viral infection, reduced availability of protein building blocks, and changes in how the cell’s machinery works) can alter the way translation occurs. Researchers know that cells from people or animal models with HD have increased amounts of cellular stress. Dr Frydman’s lab have shown in their models, that under these conditions of stress, more huntingtin proteinhuntingtin protein The protein produced by the HD gene. is made. When cells are making the huntingtin proteinhuntingtin protein The protein produced by the HD gene. by translation, they use machinery called ribosomes. Frydman and colleagues show that when cells make mutated huntingtin, the ribosomes collide and cause a traffic jam on the huntingtin message. When the Frydman lab looked at what genes were altered on the message with and without the traffic jam, they found that many of those genes were involved in protein cleanup in cells.
One protein, called eIF5A, is depleted in HD models. eIF5A is important for helping the ribosomes to clear the traffic jams, so if less of this protein is around in HD, there will be more problems making new protein molecules and clearing away the old ones. Together, Dr. Frydman’s work suggests that a whole host of molecular disruptions that result from HD occur at the level of both the RNARNA the chemical, similar to DNA, that makes up the ‘message’ molecules that cells use as working copies of genes, when manufacturing proteins. message and the huntingtin proteinhuntingtin protein The protein produced by the HD gene., each contributing to the signs and symptoms of HD we see in patients and in HD models.
Disease effects caused by huntingtin
The second session is hosted by Dr. Balajee Somalinga (CHDI) and Dr. Ali Brivanlou (The Rockefeller University) and it will focus mainly on the huntingtin message and protein and their roles in health and disease.
Early effects caused by huntingtin
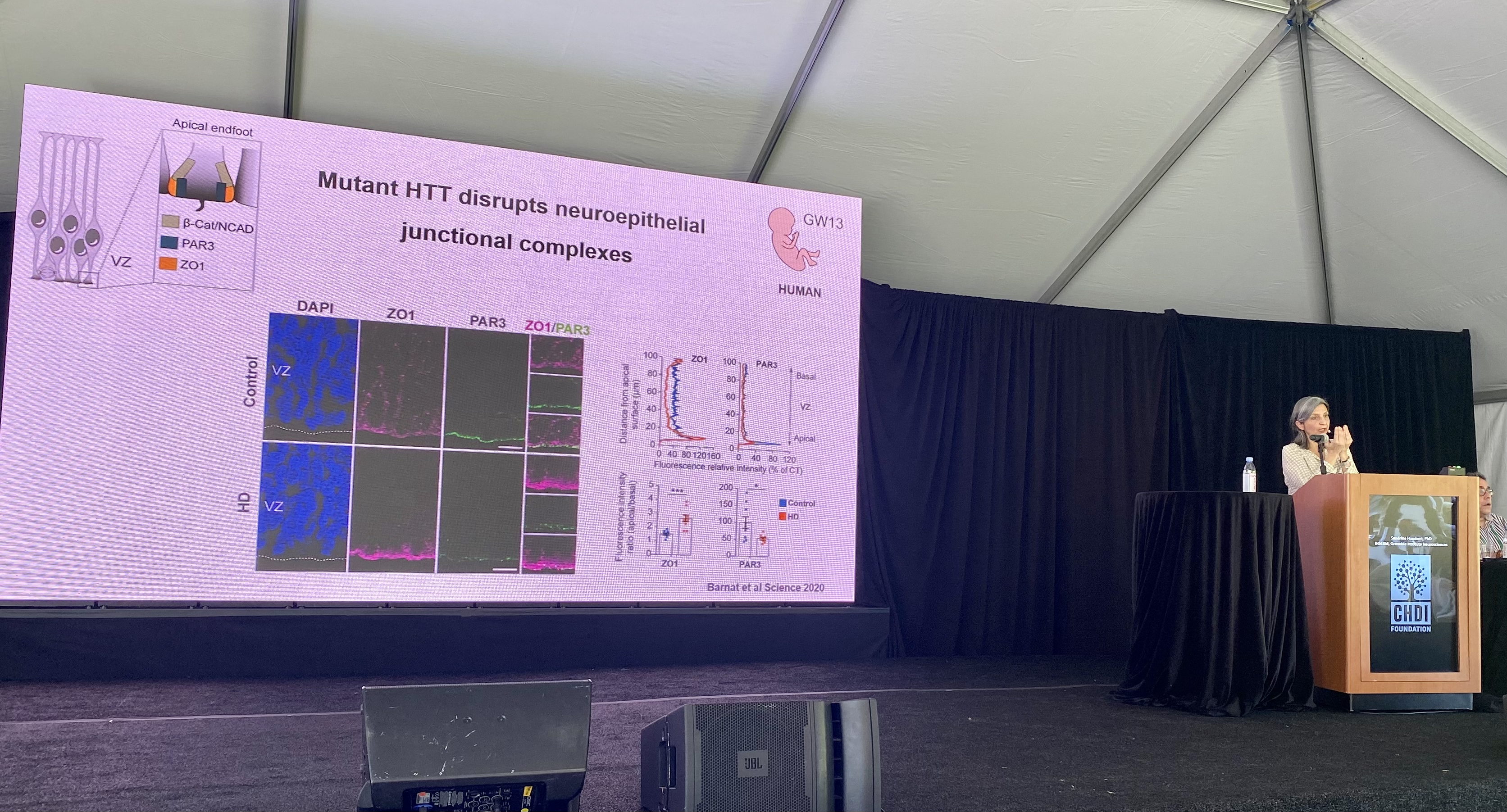
The first speaker of this session is Sandrine Humbert from INSERM, who will be talking to us about her research on the role of the huntingtin proteinhuntingtin protein The protein produced by the HD gene. during brain development.
The huntingtin proteinhuntingtin protein The protein produced by the HD gene. has lots of jobs in the cell, one of which is to move different molecules around the cell. One of the molecules huntingtin helps transport in nerve cells is BDNFBDNF brain-derived neurotrophic factor: a growth factor that may be able to protect neurons in HD, which is important for supporting the health of brain cells.
Both the normal and expanded forms of the huntingtin proteinhuntingtin protein The protein produced by the HD gene. are made by cells in the very early stages of life. The Humbert lab thinks that errors made by the expanded form of the protein in people with HD may be responsible for the symptoms they suffer later in life.
The Humbert lab has discovered that huntingtin is important for many functions in nerve cell development, including how these cells are originally formed, their final structure and how they ultimately work and connect with other nerve cells.
In HD mouse models which the Humbert lab work on, this development doesn’t happen properly which may account for the neurodegenerationneurodegenerative A disease caused by progressive malfunctioning and death of brain cells (neurons) seen later in life for these mice. We wrote about this work previously here: https://en.hdbuzz.net/290
Dr. Humbert hypothesizes that the change in the way the cells in an HD brain connect sets them up to be vulnerable later in life when HD patients would typically develop symptoms.
Her lab’s latest work continues to explore huntingtin’s roles in health and in HD, including how HD affects nerve cell growth, structure, and movement. Creating more stability within the structure of the neuronsneuron Brain cells that store and transmit information, similar to supportive scaffolding on a building, seems to have positive effects on their health later on.
In summary, it seems that even though nerve cell development is different in HD models, the nerve cells are very resilient and symptoms can still take decades to present themselves.
Huntingtin in other species
Next up is Dr. Raffaele Iennaco from the University of Milan & Istituto Nazionale di Genetica Molecolare. His work uses stem cellsstem cells Cells that can divide into cells of different types to understand how the structure of the huntingtin exon1 fragment affects function.
Dr. Iennaco works with Dr. Elena Cattaneo’s lab, where he focuses on the use of special forms of stem cellsstem cells Cells that can divide into cells of different types known as “induced pluripotent stem cellsinduced pluripotent stem cells Stem cells that have been grown from adult cells.” or iPSCs. These cells, derived from HD patients, enable the team to study the very beginning of the Huntingtin proteinhuntingtin protein The protein produced by the HD gene.. This small piece reflects just a tiny bit of the Huntingtin proteinhuntingtin protein The protein produced by the HD gene. – maybe 3% or so of the full protein. But this tiny bit plays an outsized role in Huntingtin’s jobs in the cell – particularly how it moves around the cell.
To better understand this small bit of the Huntingtin proteinhuntingtin protein The protein produced by the HD gene., Iennaco’s team determined the exact sequence of this region in 209 different animal species! This dramatically increases the numbers of species for which we have this kind of information.
The number of CAG’s across species varies quite a lot – in fish it always seems to be 4 CAGs, in lizards 5, whereas humans without HD have 17-20 CAG repeats. Why different species need different amounts of CAGs is a big mystery that Iennaco is interested in understanding. Other evidence from Iennaco’s work suggests that the huntingtin gene is unable to accept mutations – there are many fewer changes to huntingtin’s genetic code than would be expected by chance. Additional evidence for the importance of the huntingtin gene. In marmosets – go google that for a very cute monkey experience – there are actually two Huntingtin genes! This isn’t the case in any other species studied, but it suggests the power of looking at more than 200 species to find examples of rare genetic events to better understand the Huntingtin gene. Using their stem cellsstem cells Cells that can divide into cells of different types growing in the lab, Iennaco’s team could study the exact link between the length of the CAG repeatCAG repeat The stretch of DNA at the beginning of the HD gene, which contains the sequence CAG repeated many times, and is abnormally long in people who will develop HD region and the ability of those cells to develop into brain cells called neuronsneuron Brain cells that store and transmit information. These experiments help us understand the importance of all the genetic diversity identified in their sequencing studies.
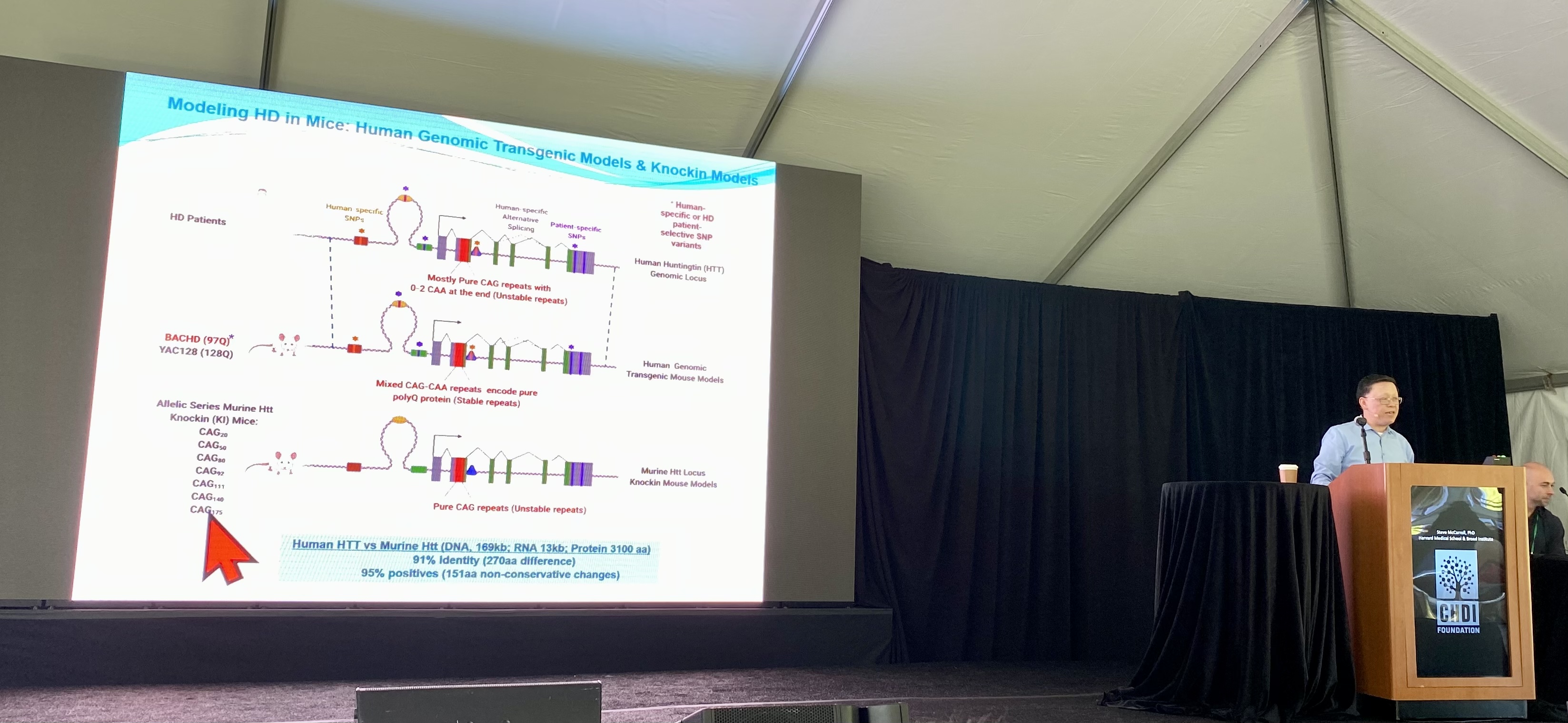
Next, Iennaco and team focused on the comparison of mouse and human Huntingtin. Strangely, while they are very similar, the human Huntingtin gene has been found to be more toxic than the mouse version, but we’ve not known why.
The team is able to coax their stem cellsstem cells Cells that can divide into cells of different types grown in dishes to begin going through the very earliest phases of brain development. This allows them to study the importance of small changes (in CAG length or across species) and to measure their impact on brain development.
Using a very cool automated system, the team took images of approximately 5,000 different mini-brains in the lab to better understand the impact of tiny changes in Huntingtin’s sequence.
Many of the aspects of new brain cell growth they measured were more impacted by human Huntingtin rather than mouse Huntingtin. This suggests that there’s something about the human sequence that sets it apart, in its ability to be toxic to newly born brain cells. The team has narrowed in on a very specific region of the Huntingtin gene that they think explains why human versions of the HD gene are more toxic than those from mice. This supports the importance of genetic studies like this in animals.
Effects caused by huntingtin in astrocytes
Next up is a talk from Prof. Baljit Khakh, from UCLA. His lab is interested in a specific type of support cell – called an astrocyte – in HD. These aren’t the most vulnerable cells in HD – that’s neuronsneuron Brain cells that store and transmit information – but astrocytes job in life is to support neuronsneuron Brain cells that store and transmit information. While astrocytes don’t die early in HD, they definitely express the HD gene, and they show a number of changes in their shape and function when they express a mutant copy of the HD gene. Khakh’s lab wants to know whether these changes in astrocytes impact HD. Khakh’s lab began their work by looking at huge data sets generated from the brains of HD patients and animal models showing which genes were turned on and off, to look for hints that astrocytes might be working poorly. This seemed to be the case. There are changes in astrocytes in the brains of HD patients, but do they matter for the progression of HD, or are they just reflecting changes in other cell types?
A very cool Huntingtin-lowering tool called a “zinc finger” can shut down expression of the mutant Huntingtin gene. We’ve written about ZFPs before at Buzz, which you can read about here: https://en.hdbuzz.net/275
The UCLA team was able to develop viruses that deliver these Huntingtin-lowering payloads to different cell types in the brain, including neuronsneuron Brain cells that store and transmit information or astrocytes. This enables them to lower the Huntingtin gene in different types of cells.
These new viruses very nicely reduce levels of the Huntingtin gene only in the targeted cell type, so the team is able to ask specific questions about the relationship between Huntingtin expression in particular cells, and HD-like symptoms in mice.
Shutting down mutant Huntingtin in each cell type rescued many of the changes found in that cell type. When mutant huntingtin is shut down in neuronsneuron Brain cells that store and transmit information (the sick cell type in HD), they saw improvements in the astrocytes – the support cells!
This is weird! It suggests that there’s some kind of feedback loop happening between sick support cells and sick neuronsneuron Brain cells that store and transmit information in the HD brain. It also shows the power of manipulating specific cell types – things aren’t always as we assume.
The team then asked the question – what happens to HD-like symptoms in HD mice if Huntingtin is lowered in astrocytes or neuronsneuron Brain cells that store and transmit information using a ZFP?
Many of the symptoms they investigated were improved by knocking down the mutant huntingtin gene in neuronsneuron Brain cells that store and transmit information, but less so when they knocked it down in astrocytes.
This is important – Khakh loves astrocytes, and wanted to understand if they drive HD symptoms. They did a very good set of experiments and find that astrocytes are changed, but that changes in neuronsneuron Brain cells that store and transmit information remain the most important factor, in light of their results.
Processing the huntingtin message
Next up is Jose Lucas from Center for Molecular Biology Severo Ochoa (CBMSO) who will be speaking about how the huntingtin message is processed and how this differs in people with HD.
The process by which gene messages are processed is called splicingsplicing the cutting up of RNA messages, to remove non-coding regions and join together coding regions.. This topic has cropped up in a few earlier talks that also looked at this process, and splicingsplicing the cutting up of RNA messages, to remove non-coding regions and join together coding regions. is thought to create the toxic exon1 fragment of huntingtin. Splicingsplicing the cutting up of RNA messages, to remove non-coding regions and join together coding regions. goes wrong in various other diseases, so understanding the similarities in this process between diseases could help answer questions about HD and the symptoms we see in patients such as loss of nerve cells. Lucas and colleagues looked to see which genes are affected by changes in the splicingsplicing the cutting up of RNA messages, to remove non-coding regions and join together coding regions. process in HD. If a gene’s message is spliced incorrectly, this will often mean that less of the full protein product of that message will be made. Scientists in the Lucas lab showed that if they switched on a gene called RBFOX1 artificially, they could improve the symptoms in a HD mouse model by helping correct the splicingsplicing the cutting up of RNA messages, to remove non-coding regions and join together coding regions. mistakes. Maybe this idea could be used to help make new medicines to treat HD?
Gene messages are also processed to remove a “tail” in their genetic code sequence which is made up of lots of the letter A repeating over and over. It turns out that in HD models, lots of messages keep their tails longer than they should, which will affect how they are turned into their protein products. One of the most affected proteins discovered in this research led the scientists to find out that people with HD have less of a vitamin called thiamine. They confirmed this by measuring the thiamine levels in the spinal fluid, showing reduced levels. The scientists are now pursuing the answers to two different questions in the clinic: Could thiamine levels be used as a biomarkerbiomarker a test of any kind – including blood tests, thinking tests and brain scans – that can measure or predict the progression of a disease like HD. Biomarkers may make clinical trials of new drugs quicker and more reliable. for progression of HD? And can thiamine treatment improve symptoms in people with HD?
While these are commonly available vitamins the Lucas group is looking at, tightly controlled clinical trials are required for conclusive answers. Hopefully we will have updates for you soon on how this possible treatment might be working in people with HD.
Controling huntingtin proteinhuntingtin protein The protein produced by the HD gene. degradation
Wrapping up the talks for today, Dr. Michael Rapé from Howard Hughes Medical Institute, University of California, Berkeley will discuss his work on how huntingtin is degraded in the cell and how it might be used to treat HD. The Rapé lab is looking for small molecules that can be used to target the huntingtin proteinhuntingtin protein The protein produced by the HD gene. so the cell’s machinery will break it down and remove it, a process known as protein degradation.
There are certain proteins in the cell that tag other proteins for degradation. So if you can control this process, you could control which proteins the cell degrades. This would be great for a disease like HD where we want to reduce or get rid of a harmful protein! One challenge with therapeuticstherapeutics treatments for brain diseases is getting past the blood-brain barrierblood-brain barrier A natural barrier, made from reinforcements to blood vessels, that prevents many chemicals from getting into the brain from the bloodstream – the selective barrier that protects the brain from harmful things in the blood. The drugs the Rapé lab are developing are small compared to ASOsASOs A type of gene silencing treatment in which specially designed DNA molecules are used to switch off a gene (like those developed by Roche and Wave) but are still big compared to most drug molecules.
Luckily scientists have shown that small molecule degraders can pass from the bloodstream into the brain which is great news for researchers looking to make degraders to treat diseases like HD.
Our cells make lots of different proteins, called E3 ligases, which are used to “tag the trash” in the cell and target it for degradation. If we could find an E3 that tags the huntingtin proteinhuntingtin protein The protein produced by the HD gene., we could harness it to develop a degrader molecule. The Rapé lab developed a screen that would allow them to identify E3 ligases that would be good targets. They identified an E3 ligase called RNF126 which seems to have all of the desired characteristics for developing of the huntingtin degrader molecule, harnessing RNF126. Next they tested if RNF126 could specifically degrade the huntingtin proteinhuntingtin protein The protein produced by the HD gene.. They found that when expression of RNF126 was increased, it led to degradation of harmful huntingtin in cells!
But these experiments were done with just a fragment of harmful, expanded huntingtin. What happens when the same experiment is done with full-length huntingtin proteinhuntingtin protein The protein produced by the HD gene. with an expanded CAG repeatCAG repeat The stretch of DNA at the beginning of the HD gene, which contains the sequence CAG repeated many times, and is abnormally long in people who will develop HD? The results replicated! Together, these data suggest that they were able to find this needle in a haystack – the perfect enzyme that binds to huntingtin to naturally allow for its degradation in cells to prevent protein aggregation that causes disease.
The next steps are to move RNF126 forward in drug development to try and identify a compound called a molecular glue which forces RNF126 to help degrade huntingtin proteinhuntingtin protein The protein produced by the HD gene.. We’ll be anxiously waiting to see what the next steps are for this exciting molecule!
Stay tuned for more updates!
That’s all for today, folks. We’re breaking for the night, but will be back tomorrow morning to continue with research updates focused on innovative approaches for HD therapeuticstherapeutics treatments!
For more information about our disclosure policy see our FAQ…


