
Huntington's disease therapeutics conference 2019 – Day 3
New insights into the huntingtin protein, and further progress in turning genetic quirks that modify the course of HD into treatments, on the final day of the therapeutics conference

Jeff and Ed report from the Huntington’s Disease Therapeutics Conference – the biggest annual gathering of HD researchers. This year’s conference is bigger and more exciting than ever.
Read About Day 1 here.
Read about Day 2 here.

Thursday morning – Huntingtin structure & function
The final day of the HD Therapeutics Conference begins with a session on the biology of the HD gene itself. It’s important to understand what this gene normally does, and how these functions are altered by the mutation that causes HD. The HD gene, called “Huntingtin” by scientists, isn’t only found in HD patients, but in every human on earth. Why do we have this gene, and what does it do to help our cells survive?
Up first, Rick Myers of Hudson Alpha, describes his lab’s work trying to understand what turns on the HD gene. Genes reside in our DNA, where they’re silent until called upon by cellular machinery, which then goes to work activating specific genes. Myers’ work might help us understand the basic biology of the HD gene, which is important, but also potentially provide new tools to lower its expression. The HD gene isn’t only found in brain cells, but many (or maybe all) the cells in the body.
Myers is part of a huge consortium of researchers, called the ENCODE project, interested in understanding how changes to the structure of DNA enables certain genes to be turned on or off. You can read more about the ENCODE project on its Wikipedia page.
In cells, DNA is chemically modified to make it sticker and more condensed, or looser and more open. It also loops around – Myers describes DNA loops interacting over very long distances. Myers’ team has identified DNA sequences, more than 100,000 DNA letters away from the HD gene itself, that control its expression. They’re basically building a map of the DNA neighborhood of the HD gene – what turns it on, and what turns it off? These huge loops of DNA around the HD gene interact in very specific ways. Since it’s so reproducible, this likely means these loops are important for how the HD gene is activated and turned on.
Up next, Prof Gill Bates of UCL, describes her lab’s work on understanding the process of “splicing” of the HD gene. Most genes – especially big ones like HD – are written in chunks of DNA, interspersed with long stretches of DNA that don’t contribute to the protein at all. So most of the DNA in the region of the “HD gene” isn’t actually part of the gene. Biology is weird! So when cells need a gene like the HD gene, they have to read each chunk – like the chapters of a book – and remove all the unimportant DNA in between. In the HD gene there are 67 of these chapters. This process of stitching together all the chapters is called “splicing”.
Bates’ lab has discovered something very funny – in mutant HD genes, a bit of the DNA between chapter 1 and chapter 2 is sometimes left in when the gene is spliced. This shouldn’t happen – each chapter is normally nicely snipped out and pasted to its neighbors. This leads to the generation of a tiny little HD protein – actually only about 3% of the whole HD protein. The Bates lab is now trying to figure out whether this tiny little HD protein could cause problems in the brains of people with HD mutations.
They’re now looking in brain samples donated by HD patients, where they see evidence of altered splicing of the HD gene, consistent with what they’ve seen in mice. But do these changes matter? Bates’ lab is working with two lines of HD mice, which have different severity of disease, to try and understand this. Mice with this splicing change are sicker, consistent with her hypothesis. Bates lab is now taking an even more direct approach, making mice which lack the extra bit of DNA that’s accidentally pasted into mutant HD genes. These mice should allow us to determine whether these splicing changes impact HD. These mice are missing most of the weird mini-HD protein, but are otherwise normal. So they should allow us to understand how much the tiny HD protein matters. It remains to be seen what this means for human HD patients!
Next Chris Ross from Johns Hopkins University introduces the topic of “post-translational modification” – that’s little chemical changes made to proteins that change they way they behave. Like how painting stripes on a car makes it go faster. Our best frenemy, huntingtin protein, can be modified in many different ways after it’s been made. We know where these modifications are, along the length of the protein, but it’s less clear what they do, why they’re added or removed, or whether they make the protein more or less harmful.
Ross’s team looked at dozens of individual modifications and tested them in cells grown in a dish. They studied whether they made the protein more or less likely to kill the cells, and found some modifications reduced harm from the Huntingtin protein. Some of these modifications, and the machines that add or remove them, could be good targets for drugs to protect neurons.

A big breakthrough last year was the structure of huntingtin was figured out for the first time – you can read more about that here. This lets clever people like Ross start to understand for the first time where exactly the modifications they’ve been studying actually sit on the protein in 3D space. But how do we turn these modifications into treatments? The first step is to figure out what change we want to produce (to make the protein less dangerous). Next is to figure out what enzyme machine puts on or takes off the most interesting modifications.
A small “ooh” went up from the audience at this nice bit of work to bring together two big areas of HD research – huntingtin structure and post-translational modification.
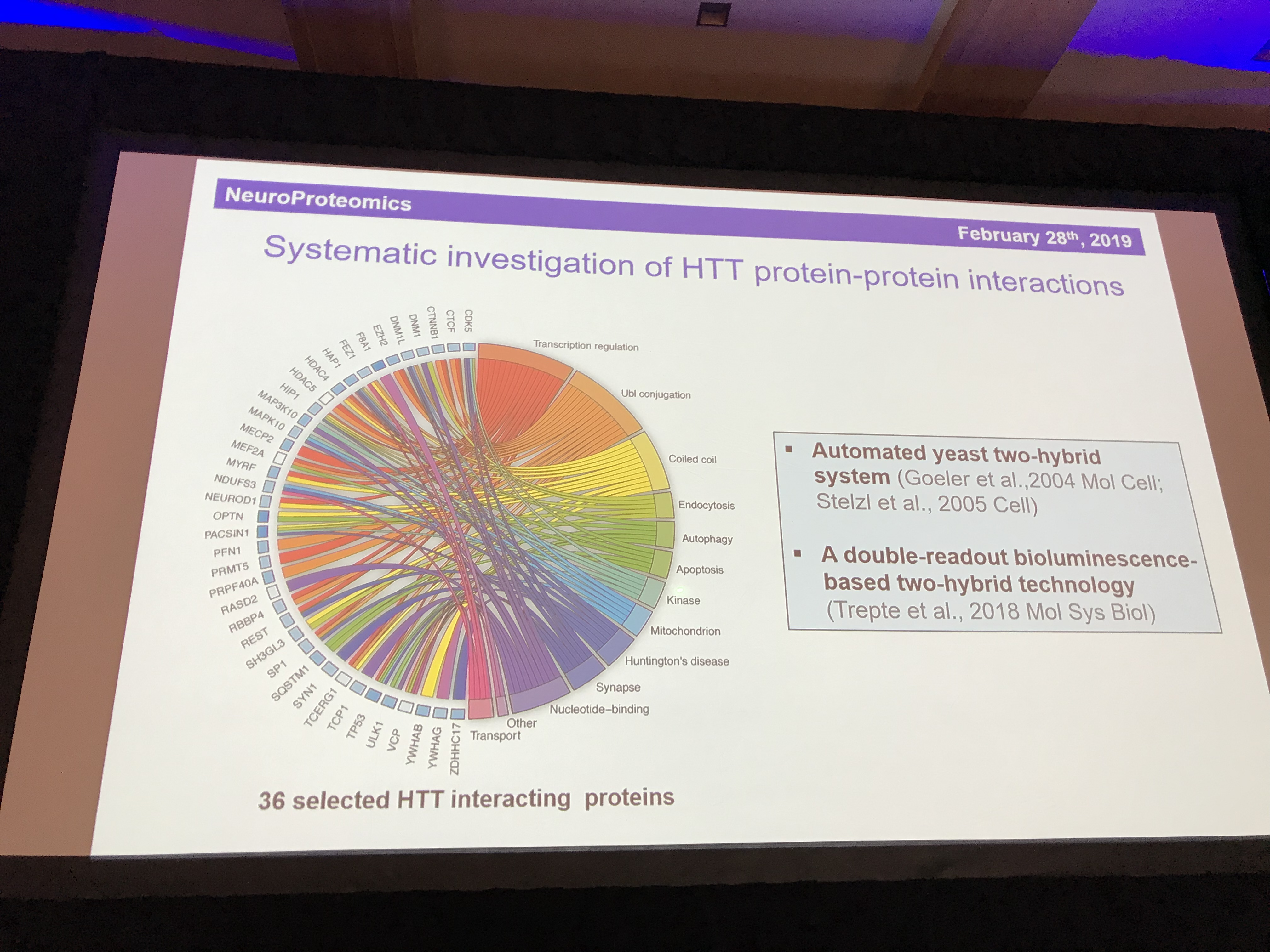
The final speaker in this session is Erich Wanker (before you ask, he’s Austrian so the W is pronounced as a V). Erich is a massive expert in the behaviour of the huntingtin protein. He uses fruit flies to study how the mutant HD protein causes cells to die. Like Chris Ross, Erich’s work has been given a big boost by the discovery of the structure of huntingtin protein last year. One of the most important things about any protein is its friends – what other proteins does it hang around with? We call these the ”interaction partners” of a protein.
Having narrowed the list of 413 interaction partners down to 36 that look particularly interesting or important, he is now looking at which of these interactions found in the scientific literature are actually real and how strongly huntingtin binds to each one. To make it more realistic, he studies the interactions inside living cells. His testing system examines the strength and closeness of the interactions to enable us to get a sense of what partnerships are most important for huntingtin. The partnerships point to important roles for huntingtin in recycling, transport, and the packaging of signalling molecules into little bubbles called vesicles. One protein, HAP1, seems particularly important. HAP stands for huntingtin-associated protein, so it’s story certainly checks out. Another important partnership is with HAP40 – but it’s not yet clear why. Mapping out the effects of these partnerships will help us understand huntingtin in its mutant and healthy forms.
Thursday afternoon – DNA Repair & Handling
The conference concludes with an exciting session to wrap up the conference focused on genetic modifiers – genes other than the HD gene we think are important for HD. Variations in these genes have been shown to make people experience HD symptoms earlier or later. We’ve talked about this idea before on HDBuzz — you can read more here.
The first speaker this afternoon is Marcy MacDonald, of Massachusetts General, a key member of the team that identified the HD gene in 1993. Since then, she has continued to work on understanding the genetics of HD.
MacDonald is discussing the work of a group of researchers, called the “GeM-HD” consortium. They’re investigating DNA variations outside the HD gene which are associated with people experiencing HD symptoms earlier or later. She says “Nature has already performed a successful HD trial”, meaning that since we’ve found people who inherited little quirks in their DNA that lead to them having later onset of HD symptoms. This is really great news because we don’t have to wonder whether it’s possible to slow HD — these folks prove that it’s possible. Now we just have to figure out how to re-create those genetic effects with drugs we can give everyone.
These genetic modifier studies begin with a large collection of HD patients, whose age of onset of motor signs of HD we know. Though this is largely defined by the “CAG size” a person inherits from their parent with HD, there is still a lot of variation in when symptoms begin. Researchers use special microchips to read huge numbers of DNA letters – 11,000,000 of them. This results in a sort of high-level map of each participant’s entire collection of DNA, or “genome”.
Having done this in 4,000 HD patients, the GeM-HD consortium found 4 different regions of the genome that were found in people who had early or late onset of HD. In fact, they found variations linked with both hastening and delaying HD. So folks that inherit one of of these variations could be predicted to experience HD symptoms either earlier or later than people who didn’t. Not only is this important for them, it’s helpful for everyone because the identify of those genes can teach us a lot about HD. Very interestingly, several of these genes are important for helping cells do a process called DNA repair. This suggests that breakdowns in DNA repair might drive the development of HD.
Now MacDonald is describing the results of their new study, with more than 9,000 subjects! This reveals several other new genes that influence HD onset, including more DNA repair genes. Interestingly, rare variations in the HD gene itself also proved to modify HD onset. We don’t say this often, but the reason is a too complicated for a highlights reel, so we’ll publish a HDBuzz story next week describing it.
MacDonald’s team is also developing new mathematical techniques to better capture the progression of HD, which they know is more complicated than a single “diagnosis”. Specifically, they’ve tried to accurately estimate how impaired people’s day to day function is in the course of HD, rather than having a single point of "onset” or “diagnosis”. They’re re-analyzing all the genetic modifier studies using these new tools to see if they can learn about which genes are important for different aspects of HD symptoms.
Up next is Peter Holmans, of Cardiff University, who is also a member of the GeM-HD consortium and has an interest in understanding how genetic changes contribute to disease. He’s addressing the conference about a type of genetic score called a “polygenic risk score”. It sounds complicated, but the concept is pretty simple – it’s a way of combining very small genetic risks into a single combined risk score. Unlike HD, many diseases aren’t caused by a single mutation in a single gene, but are influenced by many small genetic risks. Now that we can cheaply and easily measure hundreds or thousands of genetic variations we can calculate polygenic risk scores easily.
Holmans is focusing on the psychiatric symptoms of HD – things like aggression, depression and problem behaviors. They’re taking an interesting approach of looking for genetic variations that influence these symptoms independently of the general progression of HD. Using polygenic risk scores from psychiatric diseases like major depression, Holmans observes that HD patients with these genetic variations are more likely to have specific symptoms. After more work, this might enable HD families to get an idea what symptoms they’re at particular risk for developing. Since many of these – including depression – are treatable, that’d be useful information for families to have.
Up next, Hilary Wilkinson of the CHDI foundation, describes the foundation’s efforts to understand how DNA repair genes might play a role in the development of HD. Besides the HD gene itself, these new genetic leads are now the most exciting possible means to influence HD. That’s because the genetic modifier studies done in HD patients suggest that tiny changes in these gene’s sequence can significantly influence HD onset. The whole point of doing genetic screens is to identify genes that we can try to mess with using chemicals in hopes of replicating the genetic effects.
CHDI is funding and supporting a wide range of efforts to understand exactly which DNA repair genes influence the HD gene, and whether we can develop drugs to try and do this in HD patients. Using mouse models, HD researchers are able to make precise genetic edits that turn off DNA repair genes to study how they impact the HD gene and HD-like symptoms in mice. Wilkinson is describing a phenomenon called “somatic instability”, which results in the lengthening of the CAG repeat regions of the HD gene. This happens in some, but not all, cells of the body, including the most vulnerable brain cells that die in HD. Most of the genes discovered in the genome-wide studies play a role in the process of somatic instability, which has driven a huge amount of interest in the process in the HD field.
Wilkinson closed by describing a very wide range of experiments being conducted by CHDI directly, and with academic partners, to screen for drugs that might influence the process of somatic instability.
That’s all for this years HD Theraputics Conference! Be sure to have a read about Day 1 here, and Day 2 here and keep up with the conversation on Twitter.
For more information about our disclosure policy see our FAQ…


