
Huntington's disease therapeutics conference 2018 – day 3
Updates from day 3 of the Huntington’s Disease Therapeutics Conference: huntingtin protein – and lowering it

Good morning from the final day of the 2018 HD Therapeutics Conference! Two sessions today, the first focused on the protein made from the HD gene. The second includes updates on Huntingtin Lowering Trials from both Wave Life Sciences and Ionis Pharmaceuticals.
Thursday morning – huntingtin protein
Every HD patient has inherited the same mutation – a lengthening of the sequence C-A-G. This expansion happens in a gene we now call the HD gene. Genes are used by cells as instructions to make proteins – the first session today focuses on the HD protein.

Sandrine Humbert, Universite Grenoble Alpes, has long-time interest in the development of the brain, and how the HD gene and protein influence this process. To understand this process, Humbert’s lab made a mouse which lacked HD gene and protein in their brains. They discovered that cells lacking the HD gene divided and moved in an abnormal way. During brain development, newly born cells crawl towards their proper location, often climbing along ‘ropes’ formed by other cells. This process is altered when the HD gene is deleted, suggesting important roles for the HD gene in this process.
Andrea Caricasole, IRBM Science Park, is conducting a large-scale study of “post-translational modifications” of the Huntingtin protein. This refers to tiny chemical “decorations” of the Huntingtin protein. These decorations allow cells to tweak the function of proteins. The Huntingtin protein, for example, has probably dozens of these tags which are added and removed, tweaking the function of Huntingtin in response to a wide range of signals. Many of these decorations do intriguing things to the Huntingtin protein, and can even prevent mutant huntingtin protein from damaging cells. We’ve written about this before on HDBuzz. Caricasole’s team is developing very sensitive tests for individual Huntingtin protein decorations. These enable them to track which of these are changed in the course of the disease, and maybe search for ways to fix them.
Rohit Pappu, Washington University, takes a very focused approach to understanding the Huntingtin protein. His lab is developing tools to study the part of the protein whose shape is influenced by the HD mutation. Pappu’s lab uses massive amounts of computer power to try and predict the shape of the part of the huntingtin protein altered by the mutation. These techniques allow them to observe a “tadpole” shape. The shape of this tadpole has been a subject of intense debate in the HD field! Pappu’s techniques strongly support one side of this debate, which is sure to help us better understand this critical part of the huntingtin protein
Xaio-Joang Li from Emory University has developed an interesting mouse model where the huntingtin gene can be switched off in grown-up mice, in the brain, body or both. These mice don’t have expanded huntingtin genes – they just help us understand whether turning off the ‘healthy’ version of the gene has consequences. Reassuringly, when the gene is switched off, nothing bad happens in the brain. Unexpectedly, switching off the gene produced inflammation of the pancreas. It’s unclear what that might mean for patients, but current huntingtin lowering treatments aren’t expected to significantly reduce huntingtin levels in the body – just the brain. Li has also used CRISPR-Cas9 genome editing to snip out the harmful bit of the HD gene in mice. Deactivating the mutant gene in mice successfully reduced formation of the toxic huntingtin protein and the mice moved around better, too. Dr Li’s been very busy! He has also made a Huntington’s disease model pig using CRISPR genome editing. Could be useful for testing new drugs because pig brain is similar to human.
“Kochanek froze the protein and used an electron beam to take thousands of photos of it. Those were then combined by computer to produce the first ever pictures of the detailed molecular structure of the huntingtin protein.”
Ankur Jain from @UCSanDiego studies RNA – the “message molecules” generated when a cell wants to use the instructions in DNA to make a protein. Our DNA lives in the nucleus of our cells, but RNA floats freely around the whole cell. The traditional way of thinking about many genetic brain diseases is that they are caused by toxic proteins, but there is increasing evidence that sometimes the RNA message molecules produced from mutant genes can be toxic too. For instance, some RNA sequences can stick to important protein machines and prevent them from doing their jobs in the cell. One possible sign of toxic RNA is the formation of abnormal blobs of RNA seen in cells in HD and other brain diseases. Jain has discovered that he can form artificial blobs of RNA by heating it and cooling it like jello. These blobs only form when the RNA contains sticky sequences like the one from the CAG stretch in HD. It’s not clear whether these RNA blobs cause harm in HD, but they might. For instance, if the RNA is stuck in the nucleus, it can’t be used to generate proteins. Antisense molecules (similar the ones currently in HD human trials) can stick to the RNA in the nucleus and prevent them from forming blobs. Other drugs could theoretically be used to tackle the RNA stickiness issue in brain diseases too.
Exciting late-breaking talk now from Stefan Kochanek, whose lab has just uncovered the structure of the huntingtin protein! Finding out what proteins look like is a really important step in understanding how they work and how to change that with drugs. The huntingtin gene was discovered 25 years ago but the protein is big, wobbly and sticky which has made it really hard to discover its structure. One team even sent the protein into space to try to get crystals to form, but alas, no dice. Kochanek’s team has succeeded where others have failed and their results were just published in Nature. The big breakthrough was stabilising huntingtin using another protein called HAP40 (“huntingtin-associated protein 40”). Once stablised with HAP40, Kochanek froze the protein and used an electron beam to take thousands of photos of it. Those were then combined by computer to produce the first ever pictures of the detailed molecular structure of the huntingtin protein. This is seriously cool and gives us a ton of stuff to work on. One caveat though: some areas were still too wobbly to figure out the structure – including the all-important bit at the beginning of the protein that contains the mutation.
Thursday afternoon – huntingtin lowering
Big end to the day and the conference as the session on Huntingtin Lowering therapies begins. Huntingtin lowering refers to approaches that aim to lower levels of the Huntingtin protein. There’s a lot of ways to do this, but many of them target the “RNA” which is an intermediate between the information in the HD gene and the Huntingtin protein.
Michael Rape, UC Berkeley, is interested in tricking cells into destroying individual proteins in a cell. In many cases, including HD, it would be really helpful to selectively remove one specific protein. Cells have more than one protein degradation pathway – an important one uses a tiny chemical decoration called “ubiquitin” as a label. Cells recognize ubiquitin as a sort of “eat me” signal and breaks down proteins carrying them. Rape’s lab has been involved in understanding how cells use ubiquitin tags to label proteins that need to be destroyed very fast – one that might be toxic, for example. Rape’s lab has built tools that let researchers, for the first time, watch proteins go through this rapid destruction pathway. The machinery for rapid protein destruction is a powerful tool – one Rape’s lab is interested in harnessing. A recently developed technique – called “PROTAC” – allows researchers to harness the ubiquitin system to drive cells to destroy specific proteins.
Scott Zeitlin (University of Virginia) works with HD mice to try to figure out what happens when we lower mutant huntingtin, normal huntingtin or both. Bear in mind that each person inherits one huntingtin from each parent – and most people with HD have one normal and one mutant copy. Scientists call the healthy / normal protein “wild-type” because it’s the one that’s more common in the wild. These questions are important because all huntingtin-lowering therapies aim to reduce the overall amount of huntingtin protein that’s in the brain. Some, like Ionis’s drug, reduce both versions of the protein equally. Others, like Wave’s drugs, seek to lower the mutant more than the wildype protein. We think it’s likely that lowering mutant protein on its own or in parallel to the wild-type one will be beneficial – but it’s still an open question whether lowering huntingtin is safe Zeitlin has bred mice in which production of the mutant, wild-type or both proteins can be reduced after the mouse has fully grown. Zeitlin found that lowering mutant huntingtin early had a bigger effect in terms of the buildup of the protein in the brain. Similarly, early reduction of mutant huntingtin had bigger benefits on weight loss and movement skill in the mice. The same was true for reducing the production of both versions of the protein – early treatment had bigger benefits Bottom line: earlier is better when it comes to repressing huntingtin. One one test (grip strength), lowering just the mutant protein improved performance, but repressing both versions didn’t. Otherwise both approaches were roughly as effective and the key factor was how early treatment was given. Zeitlin also looked at what happens if you allow huntingtin to bounce back, and this was bad for the mice. That suggests that long-term treatment is better than short-term – exactly what you’d expect.
Jodi McBride, OHSU, describes her work using harmless viruses to deliver instructions to brain cells that help them make their own RNA-destroying molecules. One of the benefits of this kind of approach is that the viruses allow the RNA-destroying molecules to be made forever, in theory allowing a one-time treatment. McBride is studying her treatment by delivering it to monkeys, who have large complex brains that are much more similar to ours. Specifically, her team is working on delivering the virus to a part of the brain called the “putamen”. The putamen is particularly interesting, because it’s one of the most vulnerable brain regions in HD – suffering a large amount of shrinkage in people who inherit the HD mutation. McBride describes improvements in the brain surgery required for delivering the viruses, including the use of MRI to image the brain as injections happen. The viral treatment led to reductions of the HD gene RNA by about half throughout the putamen, a notable improvement on previous attempts. Next up – Mike Panzara from Wave Life Sciences, who are planning 2 trials using “Antisense Oligonucleotides” (ASOs) for HD. ASOs are short, modified, bits of DNA that enter cells and destroy a target RNA, reducing levels of the target protein
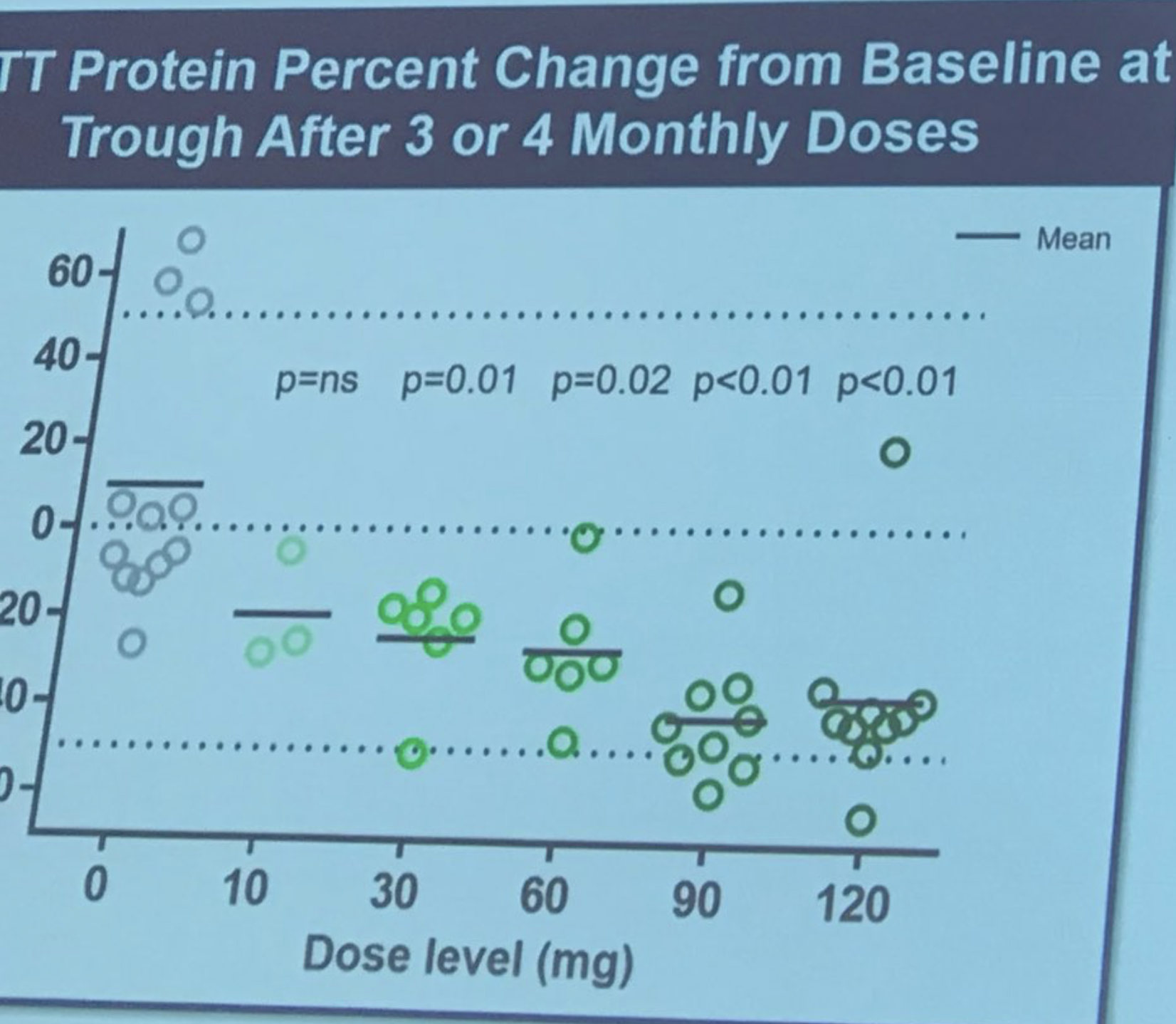
Panzara tells the crowd that Wave is currently running two trials of ASOs in HD patients. Why two? Wave’s approach relies on targeting tiny genetic variations – called SNPs, or “snips” – in the HD gene. These tiny variations don’t cause HD, they’re just part of the normal genetic variation between people – the reason we’re not all identical twins. Interestingly, these variants are found only on one of the 2 copies of the HD gene each person has. By targeting these variants, Wave’s ASOs can distinguish between the mutant and non-mutant copies of the HD gene. Wave is currently running early-stage safety studies of 2 ASOs in studies called PRECISION-HD1 and PRECISION-HD2. The ASOs used in these studies target different genetic variations in the HD gene. The trick with this approach is that people not only have to have inherited the HD mutation, but accompanying variants that allow the mutant copy of the gene to be uniquely targeted. So these trials are necessarily focused on patients carrying these variations. Wave has developed really cool new technologies to detect these variants, and determine which are on the mutant copy of the HD gene, not the normal copy. Wave conducted a preliminary study in which they were able to find targets for their ASOs in 64% of volunteers
“Spontaneous applause as Tabrizi thanks the brave volunteers in the first study, calling them “true research heroes”. ”
Next up, Anne Smith of Ionis and Sarah Tabrizi of UCL are presenting the results of a trial designed to test ASOs targeting both copies of the HD gene. This is the culmination of many years of work – Smith reminds the audience that the Ionis program started in 2005! They began with cell and animal studies, which provided early evidence that ASO treatments reduce Huntingtin protein, and make HD-like symptoms better. In 2012 and 2013 results of HD mouse model studies were published that demonstrated huntingtin lowering improved HD-like symptoms. Smith outlines the logic @ionispharma used to make the decision to use ASOs targeting both copies of the HD gene, rather than just the mutant copy. One benefit of ASOs is that they distribute widely throughout the brain. Smith shows data from monkey experiments demonstrating that after injection in the spinal fluid, ASOs distribute very widely throughout the brain. Ionis also studied distribution of even bigger animals, like pigs, finding the drug distributed very widely. Toxicity studies were then conducted, suggesting long term administration of the drug was very well tolerated (as long as 15 months in monkey studies). It’s almost impossible to sample brain tissue from patients treated with ASOs – so how will we know if the ASO did its job? Smith describes monkey studies establishing a relationship between huntingtin lowering in the brain and lowering the spinal fluid This allowed Ionis to build a very complicated computer program to predict how much Huntingtin lowering is happening in the brain and the spinal fluid, which is readily accessible by a lumbar puncture. At this point, Ionis was joined by a large pharmaceutical partner, Roche who have the resources and experience to run complicated human trials for ASOs.
Sarah Tabrizi takes the stage to describe the first human trial of Iois/Roche’s ASO treatment. This study was a “safety” study – the primary reason for doing the study was to establish if the drug was safe. The study was conducted in 9 sites in the UK, Germany and Canada. ASOs were delivered to patients by infusion into the spinal fluid in an “escalating dose”, meaning early subjects got low dose and later subjects higher. This careful ramping up of dose is done to allow safety assessments to be conducted by doctors independent from the study. This study included 46 incredibly brave volunteers, who were willing to take on some risk of being the first people to be exposed to the drug. Researchers were able to measure levels of the Huntingtin protein in the spinal fluid – which they’d previously showed correlated very well with brain levels (which, remember, we can’t measure directly).
The size of the reduction is really striking – on average as much as 40-50%! Tabrizi describes the researchers’ feelings that the huntingtin lowering may continue to improve for as much as 6 months. And here’s how much Tabrizi predicts that corresponds to in terms of brain protein lowering. Ionis has built a sort of model that enables them to make predictions about the relationship between huntingtin lowering in the spinal fluid and in brain tissues. This suggests that huntingtin lowering in brain tissue might be quite high. Patients were monitored very carefully for safety, no major adverse events were found. Tabrizi – “The drug was safe and well tolerated at all doses tested”. Success! All the subjects of the study are now on what’s called an “open label extension” – those on placebo have been moved to drug and will continue to be monitored. Spontaneous applause as Tabrizi thanks the brave volunteers in the first study, calling them “true research heroes”.
What a way to end the meeting – incredibly exciting times ahead as Roche and Ionis plan the next trial, which will be designed to determine if the drug improves HD symptoms in larger numbers of people.
Update: Ionis’ community statement on the results.
Learn more
For more information about our disclosure policy see our FAQ…


