
ASO gene silencing reaches further, lasts longer
Silencing the Huntingtin gene using ASO drugs reaches further, lasts longer and is safe. Human trial soon?
Drugs called anti-sense oligonucleotides, or ASOs, are one way of silencing the gene that causes Huntington’s disease. A new publication in the journal Neuron suggests that ASO gene silencing reaches further in the brain than other methods, lasts longer and is safe.
We have been waiting eagerly for developments in the field of gene silencing, and so we were super excited to read a major new piece of research published today in top journal Neuron. A group of researchers, led by Dr Don Cleveland at the University of California San Diego, in conjunction with the drug companies Isis pharmaceuticals, Genzyme and Novartis, have been developing anti-sense oligonucleotides (ASOs) for Huntington’s disease. So what have they been up to and what have they found?
Genetics recap

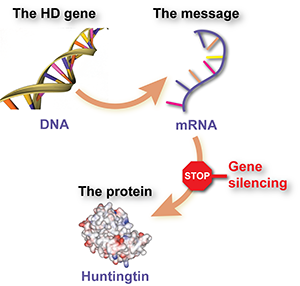
Image credit: www.biocomicals.com by Alper Uzun, PhD
The Huntington’s disease gene is just one of around 25,000 pairs of genes, made up of DNA, which carry the code for making proteins; the building blocks for the cells, which make up our bodies. There are a couple of steps between the DNA ‘code’ and the finished protein. One of those steps involves messenger RNA, or mRNA.
The HD gene is the code, which when translated makes HD mRNA. It’s the mRNA that tells a cell how to make the huntingtin protein. People who have Huntington’s disease have one normal copy and one expanded copy of the gene, so they make two different types of mRNA too.
Since the HD gene was identified, nearly 20 years ago, scientists have been trying to understand what it does, how it causes the symptoms of HD and how to effectively switch it off.
Switching off the gene
There are a number of possible methods for switching off the HD gene. Perhaps the most widely known is RNA interference, also known as RNAi or sometimes siRNA.
Another approach uses a slightly different molecule called anti-sense oligonucleotides, or ASOs.
ASOs are a bit like a cross between DNA and mRNA. They are chemically similar to DNA, but are made of a single strand like mRNA. Just like other gene silencing drugs, they are designed to stick to the HD mRNA and tell the cell to destroy it, so preventing the abnormal huntingtin protein from ever being made.
The theory behind this is that if you prevent the abnormal huntingtin protein from being made, you prevent its damaging effects on cells, and therefore reduce or delay symptoms.
In the past few months, we’ve heard good news from several groups working on RNAi drugs, but until recently the ASO researchers haven’t published as much. That just changed with this latest publication, which brings us up to date with several years’ hard work.
Human clinical trials of ASOs in other neurological diseases have already started, but the situation in HD has been slowed down by some unanswered questions.
The effect of ASOs
In this brand new work, a clever bunch of researchers have have looked at the effects of using the ASOs in 3 different mouse models of HD, and also in a monkey model (the next best thing to humans in terms of animal models), to try to figure out the answers to a number of different questions.
“ASOs treat parts of the brain that other gene silencing techniques have been unable to reach”
In the monkeys, the drug was injected into the spinal fluid – a much less invasive procedure than injecting it into the brain, and one that would be preferable for human patients.
1. What happens when you infuse an ASO, and how long does it last?
Well, they infused the ASOs into the brain ventricles (fluid filled spaces in the brain) for two to three weeks. This led to decreased levels of the abnormal huntingtin protein throughout many areas known to be important in the brain, including the striatum, which is affected most prominently in Huntington’s disease. ASOs were able to spread much further in the brain than we’ve seen with RNAi drugs.
What’s more, the levels remained low for a long time – up to three months after stopping the infusion.
2. What happened to symptoms?
Researchers are able to monitor the symptoms of animal models using tests that measure movements and behavior. The animals treated with ASOs improved compared with their untreated counterparts. Even better, improvement was sustained for a long time – and not just while the protein levels remained low. Symptoms were still better some months after the levels of abnormal huntingtin protein had returned to pre-treatment levels.
This supports the idea that the brain may only need a little assistance, to help it survive the effects of the HD gene. One prominent HD researcher, Carl Johnson, coined the term ‘huntingtin holiday’ to suggest that a short break from the harmful protein may be all that’s needed to tip the balance in favor of recovery.
3. When is the best time to give treatment?
This study suggests that early treatment is probably better.
Motor symptoms in one particular mouse model improved within a month of treatment, and continued to improve until the HD mice looked no different from normal mice. Behavioral symptoms were restored to normal within 2 months of treatment.
When older mice with more symptoms were treated, their motor and behavioral symptoms did improve, but it took much longer for improvements to be noticeable, and they didn’t gain as much back as the younger, healthier mice.
4. What happens if you block the ‘normal’ HD mRNA?
This is one of the main questions holding us back from starting trials in Huntington’s disease. We know huntingtin protein is essential for early development, as mouse embryos engineered to produce no huntingtin die before they’re born. Is it safe to switch off production of both the normal and the abnormal huntingtin protein in adults?
Thanks to this work, and the work of other gene silencing researchers, we are getting closer to an answer. Switching off normal HD mRNA for up to 3 months in healthy monkeys was well tolerated. In the animal models of HD, switching off both the normal and the abnormal mRNA didn’t change the amount of recovery and didn’t have any bad effects.
The only possible sticking point now, is that humans may be more sensitive to having less huntingtin than any animal we could test the drugs in. Only a trial with patients will tell us for sure.
This is all good news
We now have proof that ASOs treat parts of the brain that other gene silencing techniques have been unable to reach. Not only that, but a short-term infusion with ASOs was enough to delay the progression of symptoms in Huntington’s disease animal models. And the reversal of symptoms lasts much longer than expected, even after levels of the abnormal huntingtin protein return to normal.
What happens next?
We know that people who carry the expanded HD gene can remain perfectly healthy many years, despite producing the abnormal huntingtin protein from birth.
Perhaps a one-off treatment with ASOs, or treatment once a year, will be enough to ‘reset the disease clock’ by blocking the production of the huntingtin protein for long enough to let cells clear the build-up. The next stage in this group’s research will be to look at how long a single ASO injection will last.
This work also suggests that infusion into the spinal fluid might be good enough for ASO drugs. That’s no walk in the park, but is relatively straightforward compared to fitting tubes and pumps to get drugs directly into the brain.
We also need to figure out how much ASO will be needed to produce an effect in humans, when and for how long it should be given, and be prepared for unexpected side effects.
But it seems we are edging ever nearer to human HD gene silencing trials. And with different several groups all on the brink of trials, and eager to be the first to test their drug in patients, it’s truly an exciting time for gene silencing in HD.
Learn more
Sources & References
For more information about our disclosure policy see our FAQ…


