Huntington’s disease therapeutics conference 2020 – Day 1
HDBuzz reports from the annual Huntington’s disease therapeutics conference in Palm Springs


Our new writers Rachel Harding and Sarah Hernandez report from the Huntington’s Disease Therapeutics Conference – the biggest annual gathering of HD researchers.
Tuesday morning – HD genotype and phenotype
Good morning from sunny Palm Springs! We’re excited to be here for the 15th Annual HD Therapeutics Conference. This year in addition to Ed and Jeff, we’re joined by HD Buzz’s newest writers – Dr. Rachel Harding and Dr. Sarah Hernandez. Joel Stanton is compiling our live twitter output into our daily posts.

Session 1 is called “Genotype and Phenotype”, talks focused on how the HD mutation (genotype) changes HD symptoms (phenotype). First up is Dr Seth Ament, describing his lab’s work on trying to map the first changes that happen in the brain cells of mice carrying the HD mutation. The cells in our body, including our brain, have DNA that encodes more than 20,000 genes. Which genes are turned on or off in a given cell determines how that cell works. Ament’s lab has been studying this shift in what genes are expressed, and strives to understand the specific factors that make HD cells express different sets of genes, in hopes that it might be fixable.
First Ament describes his work mapping the locations where the huntingtin protein, the product of the HD gene, sticks to DNA. The most obvious way that huntingtin could change what genes are turned on or off is by sticking directly to DNA. In fact, the huntingtin protein sticks to different parts of the genome in HD mice compared to regular mice. This suggests huntingtin may be doing something directly to DNA that is important to understanding HD.
In mutant HD mice, huntingtin sticks to DNA in places where there’s lots of action – genes being read out and used. This suggests mutant huntingtin might be doing something unique in areas where genes are being actively used. Could this change in huntingtin sticking to DNA in different areas help explain how cells with mutant huntingtin get their sets of cells slightly scrambled?
In fact, the regions that huntingtin sticks to in mutant mice contain the very genes that are changed in HD cells, suggesting this might be true. Ament’s team found a surprising relationship: they can predict how active or inactive a given region of DNA is by how well the huntingtin protein sticks to it.
Next up, Ament describes his lab’s efforts in mapping which genes are changed in HD brains. Amazing new technologies allow researchers to map genes in individual cells. Ament’s lab works with these techniques in NIH’s BRAIN Initiative. Ament’s lab used these new techniques to examine changes in more then 13,000 individual cells from HD mouse brains. There are a number of different types of cells in the brain, and Ament’s approach allows him to map changes in each of those cell types separately. This gives a much clearer picture than just mushing them up and analysing together. These results paint the way to a much more refined understanding of what exactly is happening in each type of cell – which might help us understand how to treat each cell type individually.
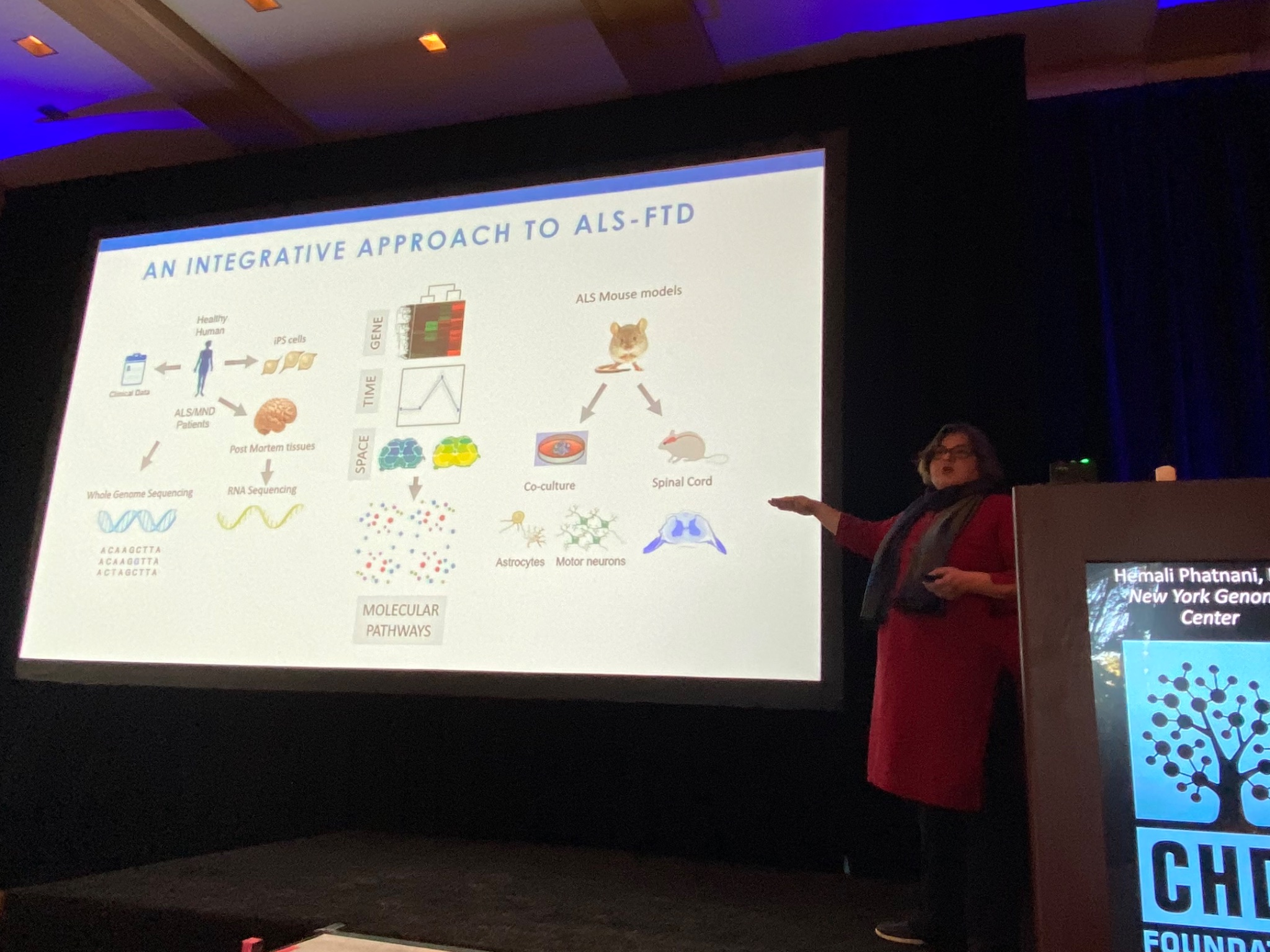
Up next is Hemali Phatnani of the New York Genome Center, who describes her work understanding the changes in the brain and spinal cord of patients with ALS (Lou Gehrig’s disease), or another related disease called FTD. Phatnani works with a large team of ALS clinics to get access to rare samples donated by ALS patients. These are analysed and the data made available immediately to researchers around the world, a great model of open science! Like HD, ALS is a complex disease; different cell types in the brain undergo different changes during the course of the disease. Phatnani’s team has helped develop new methods to map changes in cells from ALS brains. Their techniques allow them to study cell specific changes, and their data are available for any researcher (or curious non-researcher) to explore at als-st.nygenome.org.
Next is Sumanjit Jayadev from the University of Washington Medical Center. Jayadev is interested in studying a particular type of brain cell called microglia. Microglia play a role in the progression of HD, and getting rid of microglia in mouse models can help with HD symptoms. Scientists have known for a while that HD causes an inflammatory response, and Jayadev is interested in which brain cells (other than neurones) are playing a role in this inflammation. Genes involved in inflammation are risk factors for Alzheimer’s disease (AD). Jayadev is looking at which which cells in the brain these genes are switched on, to identify risk for disease using a cool technique which provides data at the level of single cells. Examining these changes in single cells has allowed researchers to identify subtypes of microglia, and looking at these subtypes identified one particular type of microglia only present in AD. Jayadev is working on this project with the folks at Sage Bionetworks who are experts in open science and open data in biomedical research. With all the data generated, then can monitor disease progression of AD by looking at which genes are switched on where and when. This can help define different populations of patients which helps clinicians and researchers understand how the disease process works. If we can apply this to HD, then perhaps in the future by knowing the age and CAG length of a patient, doctors could make informed decisions about how to treat that specific patient.
The next talk is by William Yang, a researcher from UCLA who studies HD. The Yang Lab generates lots of really large datasets from different HD mouse models, looking at how different genes are switched on, which proteins are present in different cell types, and bring all this data together to compare HD to controls. In these big data sets, scientists can search for patterns and correlations which might indicate how some genes work together in HD mouse models. These patterns can be mapped using computational methods to understand how certain cell types in the brain contribute to changes in the gene expressions they observe.
In this talk, Yang is focusing on the analysis that was done using control mice without HD, where his team showed that this technique identified important functions and which cell types contribute to these functions. When data from the HD mice model is overlaid on the map, they find that genes that regulate sleep/wake cycles and DNA repair and changed – confirming findings researchers have reported before. This map can also be used to test lots of new theories researchers have about HD – a welcome tool!
Tuesday afternoon – Somatic instability
The afternoon session focuses on the process of somatic instability. Simply put, somatic instability occurs when long repeated DNA sequences are unstable in certain cell types.
First up this afternoon is Darren Monckton from the University of Glasgow. He is interested in studying how somatic instability can drive Huntington’s and how targeting instability could be a good strategy for making new drugs for HD. We have known for a while now that the CAG-length can vary in tissues with some cells having much longer lengths than others. This is similar to what we see in other expansion diseases such a myotonic dystrophy, The variation in the CAG-repeat length gets larger as patients get older, suggesting there is more instability. Similarly the longer the CAG-repeat, the greater the variability and therefore more I fired instability. It is worth noting that this doesn’t mean your CAG number gets longer overall as you age, this will stay the same. it just means that in a few cells, the CAG-repeat length can sometimes increase.
CAG encodes for the amino acid glutamine, which is why you will sometimes hear HD called a polyglutamine disease. But glutamines can also be encoded by CAA, so although the DNA is different, the protein which is made will be the same with a CAG substituted for a CAA. This happens, for example in the HD gene, where a long “C-A-G” repeat grows longer in come cell types. There is a more detailed post on HDBuzz about this here.
A recent interesting finding showed that the presence of CAA in some places instead of CAG is better at maintaining repeat length stability. The next interesting question becomes, what is driving these changes that cause somatic instability? Evidence is no pointing to DNA repair, a recent hot topic in HD research.
Identifying specific “drivers” (or genes) of this instability could have therapeutic implications for HD. Researchers are now working to fogure out which of these drivers are most important in patients, as well as working out which are suitable for targeting with a new therapy or medicine. Now the researchers are looking to see how the instability or changes in the CAG-length, varies for a specific patient over time, all thanks to those patients who contributed samples and data to EnrolHD.
If we can monitor and track HD progression by measuring instability with a simple blood test, this could be a less intrusive way for doctors to monitor patients disease progression and suggest the best way for how they might be treated.
Next up is Karen Usdin from the NIH who has been looking at somatic instability in mouse models of a different disease which affects the nervous system called fragile X. Like HD, fragile X is a repeat expansion disease, but instead of CAG-expansion, this disease has a CGG expansion. We can learn a lot from other expansion diseases as scientists believe that there are lots of similarities in the drivers of the diseases. Also like HD, the CGG repeat that causes Fragile X is affected by somatic instability, and genes involved in DNA repair influence this process. Uddin is finding, at least in Fragile X mice, that altering levels of DNA repair genes prevents the expansion of the CGG repeat and even removes some of the repeats! It’s super to hear from scientists at this meeting from outside the field of HD, who share a lot of interesting ideas and knowledge which might help push HD research forward faster!
The last talk of the session is from Ravi Iyer of CHDI, discussing one of the drug discovery programs scientists are interested in. One of the goals of this drug discovery program is to identify small molecules that will stabilise CAG expansion in HD. CHDI is working with lots of different companies to discover small molecules using lots of different techniques – collaboration on these tricky projects helps push things forward faster.
One of the ways in which they identify small molecules that might work, is by looking at detailed models of the structures of molecules which they want to target, such as proteins involved in DNA repair. The best part about programs that seek to identify small molecules in that they could be taken as a pill if they were shown to be effective treatments for HD. While the prospect of small molecule treatment is very exciting, researchers have to be extra cautious that the small molecules they want to use aren’t having unexpected side effects.
Making small molecules which might be medicines for HD is an exciting project, but we are still way off learning whether this will be successful. A big team of scientists lead by CHDI are working hard towards which and we’re excited to see updates in the coming meetings.
Now we’re hearing from Brian Bettencourt from Triplet Therapeutics, one of the many companies working in this area, who will be telling us more about therapeutically targeting somatic instability. One of their goals is to stop somatic instability of the CAG expansion and delay the onset of HD, hopefully to an age that’s so old it’s not a realistic lifespan! This has been such a promising area of research that scientists have to prioritize which molecules to work on first. This allows them to efficiently work toward their goal of developing promising therapeutics for HD as fast as possible. After prioritizing candidates for safety and low risk, Bettencourt’s group developed molecules that target 8 different genes. While this is a large number of targets, it’s a small enough number to get done relatively quickly. Again, this research is part of a larger collaboration, as Bettencourt is working with HDBuzz’s Jeff Carroll to study these different potential therapeutic targets. Bettencourt’s group should be reporting back on some of these studies next year, so stay tuned to see if targeting somatic expansion can be used to treat HD.
The last speaker of the day is HDBuzz’s very own Jeff Carroll, who will be wrapping up the session on somatic instability. One of the things Carroll is interested in is understanding the effect of lowering huntingtin on non-brain tissue, such as the liver. Interestingly, lowering huntingtin reduces somatic instability specifically in some tissues, but not others. The Carroll lab wanted to understand these findings in more detail so are collaborating with Sarah Tabrizi to look in human neuron cells. Huntingtin lowering in a different mouse model that doesn’t have HD (but has ataxia, a different CAG repeat disease) shows that somatic instability is also reduced there, perhaps suggesting that the huntingtin protein in general has a role in somatic instability. If huntingtin is playing a role in genome stability, and how well our DNA is maintained, the Carroll lab is keen to work out how that might be happening and how this would affect HD patients. This is a very new observation so folks are excited to explore this finding!
And that’s all for Day 1! Stay tuned for our write ups of day 2 and 3 and keep up with the conversation on Twitter.
For more information about our disclosure policy see our FAQ…