An early role for the Huntington's disease gene – but don't believe all the headlines
A surprising new paper sheds light on the role of the HD gene early in development. Should we worry?

New hints are emerging about the normal role of the gene that causes Huntington’s disease. A recent report uses cutting edge techniques to study this question in cells growing in the lab. We’ll help separate the fascinating new science from some scary-sounding headlines.
Cool science, shame about the reporting
You might recently have seen some ominous headlines like “Huntington’s ‘first domino’ may fall before birth” and even suggestions that huntingtin-lowering drugs “may actually do more harm than good”. These stories were sparked by the recent publication of an intriguing study from Ali Brivanlou and colleagues at Rockefeller University. Bottom line: the science is interesting, but it doesn’t really have any implications for huntingtin-lowering drugs being tested in HD patients.
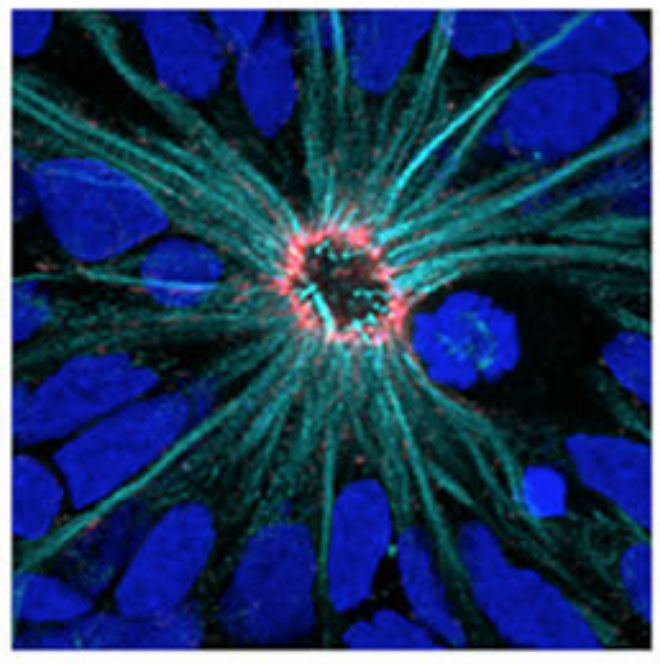
Image credit: Ruzo et al
A new tool for studying HD
Brivanlou is a developmental biologist – someone who studies how a single fertilized egg divides and eventually grows into a fully-formed adult. Developmental biologists often study the very earliest stages of development by watching cells grown in a dish in the lab.
More recently, Brivanlou’s lab has been conducting studies of the gene that causes HD, and the protein cells make using the instructions in that gene. His lab has great technical expertise at a few technologies that are key to understanding these most recent results.
First, like many labs around the world, Brivanlou’s team is using a radical new technique called genome editing with CRISPR/Cas9 to modify the DNA of cells.
Second, Brivanlou’s lab is amongst the world’s best at studying cells from early embryos as they become cells of a mature animal. Brivanlou, in fact, is well-known for recently growing human embryos in his lab for 13 days, much longer than the previous record of 9 days.
Very early in human development, all the cells are very similar. Scientists call cells like this stem cells. As time goes on, stem cells take on different characteristics – becoming skin cells, or muscle cells or brain cells, for example. What’s special about stem cells is that they have the ability to become any of these different types of adult cells, if they’re exposed to the right conditions.
The new study from the Brivanlou lab combines these techniques, by modifying the DNA of very early, unspecialized, human cells called embryonic stem cells (or ESCs). Specifically, they modified the DNA of these cells to mimic the mutation that causes HD.
Working initially with cells with normal HD genes, Brivanlou’s team used genome editing to make a few different edits. First, they deliberately expanded the CAG sequence in the normal HD gene to make it longer, like it is in people destined to develop HD. They then made another big edit, which removed the HD gene entirely.
This generated some very cool tools – stem cells that are genetically identical, except that some of them have the HD mutation, some of them have a normal HD gene, and some of them have no HD gene at all.
This is a very neat system for studying the very early effects of the normal huntingtin protein, and the mutant version produced by the HD mutation.
What did the study find?
Using the new cells they’d created, Brivanlou’s team set out to study the very earliest stages of brain development. This normally happens deep inside an embryo, but researchers can mimic the process by allowing cells to grow in dishes in the lab, where they organize into structures that somewhat resemble the early developing brain, called neural rosettes.
The rosettes showed some interesting changes when the HD gene was mutated. The shapes formed by the cells were altered in cells with the HD mutation. Interestingly, similar shape changes were seen in cells that had no HD gene at all. This finding is interesting because it suggests that the things that go wrong in stem cells when the HD gene is mutated may be similar to what goes wrong when the gene is missing altogether.
“We don’t know if these subtle changes happen in real brains, where there are a lot of mechanisms for correcting errors in development.”
Turning to the individual cells in the dish, Brivanlou’s lab discovered another interesting feature. They found that new baby brain cells called neurons didn’t all divide normally when they had the HD mutation. About 7% of the time, the process of division that separates two new cells from each other went wrong, and the cells ended up doubled.
Again, a similar kind of problem was observed in cells missing the HD gene altogether: occasionally (about 5% of the time) these cells also failed to divide completely.
To researchers trying to understand HD, this is fascinating – it seems that, in these stem cells in the lab, the way cells behave when they have the HD mutation is similar to how they react if the HD gene is completely gone. Brivanlou’s lab did some additional work to better understand exactly which stage of cell division was not happening correctly in cells with HD mutations, or no HD gene at all.
A rosette by any other name… is still not a brain
The findings described above are the complete set of findings of the study. All the work was done in cells growing in a dish in the lab, either in isolation or in a rosette structure.
The science itself is cool, and done very well, and raises important questions about how much of what we see in Huntington’s disease might depend on subtle changes that may occur in the HD brain.
But you’ll notice that we haven’t told you that anything similar happens in the brains of people carrying the HD mutation, or even animal models of HD like mice or rats. We haven’t told you that because it’s not part of this study – those questions were not asked. So we don’t even know if these subtle changes happen in real brains, where there are a lot of mechanisms for correcting errors in development.
Does the brain develop normally in HD?
So, should we be worried that brain development goes badly awry in people carrying the HD mutation? We think not, based on the best evidence that we know of so far. This evidence actually comes from people with the HD mutation, not isolated cells grown in a dish.
First, and most importantly, no one has ever found any significant changes in the personality, intelligence or mood in people who carry the HD mutation who are very far from predicted disease onset. This means that if it’s true that carrying the HD mutation leads to significant problems when the brain is developing, they’re so subtle that they’re impossible to detect with even the most sensitive tests.
Second, the TRACK-HD study followed a large number of people with the HD mutation for 3 years. During that time, the participants were examined very intensely, including the creation of very detailed brain maps. One group of TRACK-HD volunteers carried the HD mutation but was many years before expected symptom onset. Their brain scans tell us what a brain looks like after going through development with a mutant HD gene.
Crucially, almost nothing was different about the brains of those people, compared with healthy controls who didn’t have the HD mutation. Very sensitive brain imaging tests revealed tiny changes deep in the brain, but not in the regions where Brivanlou found abnormalities in his stem cell models.
In terms of symptoms, this far-from-onset group had no increases in apathy, no increases in problematic behavior, no changes in irritability, no change in walking or movements, no changes in emotional recognition, no changes in eye movements or the ability to hold a flexed muscle. They also had no changes in total brain volume. In short, the brains of people 10 years before the predicted onset of HD look and act almost entirely normally. What’s more, doubled-up cells like Brivanlou’s team reported have never been seen in the donated brains of HD patients.

It’s possible, of course, that there are very subtle changes in the brains of people carrying the HD mutation that are missed by these tests. But extensive studies done in real patients don’t support the idea that the development of these brains went badly wrong in some way.
Do we need to rethink how to treat HD?
The most dramatic interpretation of this new work from the Brivanlou lab is that the HD mutation causes changes in brain development that directly lead to HD symptoms many years later. According to an article published by futurity.org, Brivanlou appears to favour this interpretation:
“We should rethink our approach to treating Huntington’s. … by the time a patient is displaying symptoms, it may be too late to medicate. We need to go back to the earliest events that trigger the chain reaction that ultimately results in disease so we can focus new therapies on the cause, not the consequences”
However – importantly – Dr Brivanlou has since confirmed to HDBuzz that he was misquoted in the Futurity article and in fact did not talk to Futurity at all. See the note at the end of this article.
Scary stuff!
And yet, remember that none of these effects have been proven to occur in the intact brain. Even if they did happen, no one has proven that they have any link to the development of the disease. Much more work would be needed before we could understand if this was just a curiosity particular to the model system, or whether it’s something that actually drives the malfunctioning of brain bits that causes HD.
Safe or sorry?
So do we have to wait years to find out whether these fears are correct? We think not, based on a very large number of animal studies of huntingtin lowering.
In these approaches, drugs that reduce the activity of the HD gene are given to adult animals. If HD is a disease of altered development, these treatments couldn’t possibly work – it would be too late to stop the first domino from falling.
But time and time again, in experiment after experiment, many labs have found that lowering activity of the HD gene leads to improvements in HD-like symptoms in animal models. These studies have been summarized in a number of review articles; readers interested in a closer look can find them in the links section above. This is the whole justification for the ongoing huntingtin lowering treatments happening in HD.
In short, if HD is a disease of altered brain development, then treating adult animals would have no effect. But it has a huge beneficial effect. The idea that changes in brain development are the major cause of HD later in life is just not true, at least in animal models.
What about the suggestion we’ve seen in a couple of second-hand reports, that Brivanlou’s work might mean that huntingtin lowering is dangerous – because in his stem cells, removing the HD gene altogether produced similar effects to having a mutation in the gene?
“If HD is a disease of altered brain development, then lowering huntingtin adult animals would have no effect. But it has a beneficial effect. The idea that changes in brain development are the major cause of HD later in life is just not true.”
Well, the huntingtin-lowering experiments in HD animals also disprove that. If huntingtin lowering was dangerous in the way those reports imply, the animals would have got worse, not better. What’s more, huntingtin-lowering drugs have also been tested in many healthy animals including dogs, pigs and monkeys – and no ill effects have been seen. Reviews of these studies are also linked above.
Finally, as reported in December, a group of magnificent volunteers recently showed us that treatment with a huntingtin-lowering drug, IONIS-HTTRx, was safe over a three-month period. Those same volunteers are now starting to tell us about safety over longer periods, in an extension to the HTTRx study. And we expect the announcement of detailed results from the first HTTRx study very soon.
Unexpected problems could emerge – that’s why these drugs are still in trials. There are a few studies worth mentioning that should keep us on our guard. One of these, covered in a previous HDBuzz piece, suggested that completely wiping out huntingtin in adult mice could cause neurological and behavioral problems.
Another study by the Frisen group found that, in contrast to the old adage that we can’t make new brain cells, new neurons are found in the striatum (the brain area normally affected by HD). Further, the new-comer neurons are absent in advanced stages of HD. This suggests that the huntingtin protein may play a role in so-called adult neurogenesis. If so, we can’t dismiss the risk that removing huntingtin may affect this process.
In short, we have reasons to be cautious, but this new study is not nearly as frightening as the headlines might lead you to believe. As with any new drug, we need to proceed carefully. The huntingtin-lowering drugs being tested are dose-dependent and reversible, and, unlike studies that completely remove huntingtin, would never remove the protein completely. The clinical trial program has been designed to detect any possible problems and react accordingly.
Take-home
In the opinion of HDBuzz, this new work from the Brivanlou lab is fascinating, beautifully-done science. It shows that, in isolated cells in a dish, there are similarities between what happens when the HD gene is mutated, and when it’s absent. This raises really important questions about what the normal gene is doing, and how HD interacts with this normal role.
But we think it’s wrong to interpret these data as meaning that HD is fundamentally a disease of altered brain development. There’s very little support for that idea from careful studies of real patients, and many more experiments are required before such a claim is made. What’s more, there is no basis for using this work to try to predict whether huntingtin lowering might be beneficial or dangerous – and so far, the research that is relevant to that question points to it being safe.
All science proceeds by finding unexplained facts and figuring out the answer to them. New ideas are good – but we shouldn’t be tempted to read more into them than is justified. In the words of Carl Sagan, “It pays to keep an open mind, but not so open your brains fall out.”
Update, 27th February 2018. We reached out to Dr Brivanlou for his comment before publishing this article but he’s been understandably busy. He has now clarified a number of important points by email. He says he never spoke to Futurity, and never said anything about his work having relevance for huntingtin-lowering drugs. He has asked Futurity to issue a correction. Dr Brivanlou’s advice: “my view is that patients should follow the advice of their physicians. What I do is basic science.”
We have edited this article to make it clear Dr Brivanlou was misquoted by Futurity, and we thank him for his clarification.
Learn more
Sources & References
For more information about our disclosure policy see our FAQ…


