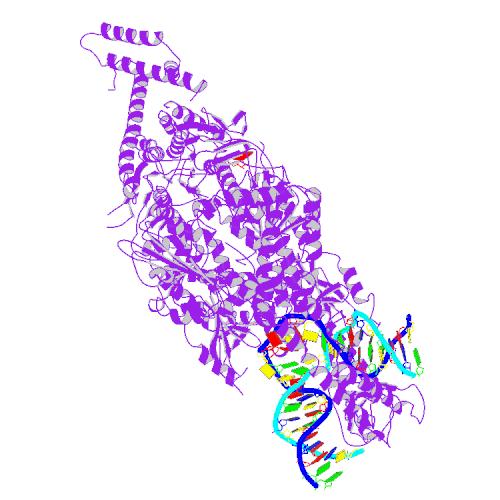
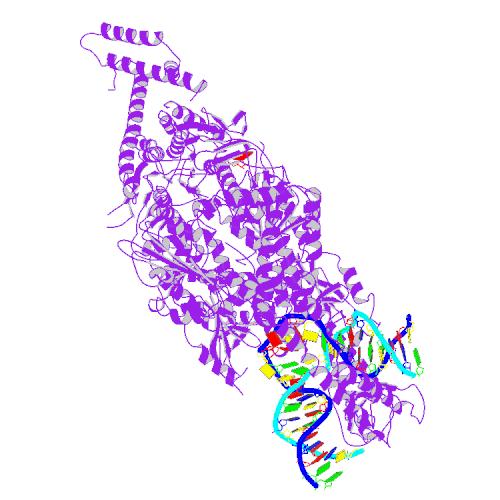
A DNA repair protein modifies the stability of long CAG tracts in the Huntington's disease gene
DNA repair is a critical process to cells, but errors in this process might explain 'repeat instability' in HD
The protein MSH3 performs an important function in cells, searching out and helping to repair genetic mistakes and damaged DNA. However, a new study from scientists at The University of Toronto, Hospital for Sick Children, suggests MSH3 may aid Huntington’s disease progression by increasing CAG repeat instability in the brain.
The problem of genetic instability
The basic cause of Huntington’s disease is well established. A repeating segment of three building blocks of the genetic code Cytosine-Adenosine-Guanine (or more simply, ‘C-A-G’) near the beginning of the HD gene is excessively long in people who develop the disease.
The CAG repeat shows great diversity throughout the population, even in unaffected individuals. Normal repeat lengths can range anywhere between 6 and 36, while repeats longer than 40 invariably lead to Huntington’s disease.
But the CAG repeat length has othert, predictive properties. Not only can the number of repeats determine whether or not a person will develop Huntington’s disease, but it can also roughly foretell the age at which disease symptoms may begin to emerge. The longer the repeat length, the earlier HD onset, on average.
In families who carry the mutation, HD sometimes displays ‘anticipation’ – a technical way of saying that disease onset can get earlier with each successive generation. Scientists have discovered that this anticipation can be explained by a biological event called ‘genetic instability’.
Instability refers to a tendency for repetitive stretches of DNA to grow throughout an individual’s lifespan. This can cause, for example, the C-A-G region in the HD gene to get longer.
For reasons that are not understood, genetic instability happens more often in some tissues and cell types than in others. For example, while repeat lengths are quite stable in the blood (where samples are collected for HD genetic testing), they often expand in sperm cells.
This genetic instability explains why anticipation occurs in HD. Repeats tend to get longer, on average causing children to have earlier onset than their parents. Because this expansion is especially common in sperm cells, expansions are more likely in HD genes inherited from fathers than mothers.
The inter-generational effects of genetic instability have been known for quite some time. However, newer studies are shedding light on how it may also affect disease prognosis within individual patients.
Instability in the brain
Brain damage in Huntington’s disease has a specific pattern: not all parts of the brain degenerate to the same extent. The parts of the brain that are most vulnerable to dying in HD are called the striatum and cerebral cortex. Interestingly, scientists have discovered that these brain regions are also the areas that show the most significant levels of repeat instability.
Since repeat length is so closely associated with HD age-of-onset, the lengthening of the CAG repeat within these brain regions could possibly explain why they are selectively lost in the disease. To further support this idea, HD patients who display the most severe brain damage are the ones with the highest levels of CAG repeat instability.
These findings raise an important question. Why do some HD patients have higher levels of genetic instability in the brain? Which problem comes first, does more brain damage cause increased instability, or does instability cause worse brain damage?
In an attempt to tackle this issue head on, a team led by Dr. Christopher Pearson at the Hospital for Sick Children in Toronto searched for genes that could control differences in genetic instability from one person to the next. In this quest, one gene, called MSH3, emerged as the prime contender.
“Unfortunately, one by-product of the repair process is that additional CAG repeats can be erroneously introduced to the sequence. Like a landslide, the more repeats that are added, the greater the original problem becomes. This is the root of genetic instability.”
The evidence
To look for genes that might control genetic instability, the researchers added a small but toxic fragment of the Huntington’s disease gene into two different strains of mice. Different strains of mice are sort of like people from different families, or different breeds of dogs – although they are all mice, they have distinct genetic backgrounds.
The researchers discovered that Huntington’s disease gene instability occurred in one strain of mice (called B6), but not the other (called CBy). From previous research on genetic instability, conducted by the Pearson lab and others, the researchers suspected that the dramatic difference between the mice might boil down to variances in one particular biological process, called mismatch repair, and more specifically one of its major players, the MSH3 protein.
To test if genetic differences in MSH3 were causing the alteration in repeat instability, the scientists transferred the MSH3 gene from CBy mice (which previously did not show CAG repeat expansion) into the B6 strain, and vice versa.
The results were dramatic. Flipping the genes between the mouse strains caused a complete reversal of repeat instability. The CBy mice, which had previously been immune to increases in repeat length, were now the strain with the highest levels, while the B6 strain showed little repeat instability at all. The effect followed the MSH3 gene!
To determine what might be causing the discrepancy, the researchers looked at the genetic sequences of the MSH3 gene from both mouse strains. Within the CBy strain, a single mutation was identified that was found to have a significant effect on function of MSH3. This single mutation caused the MSH3 protein to become unstable, and quickly degraded by the cell. As a result, any MSH3 made by the cells would be quickly broken down and recycled, reducing its overall levels substantially.
These findings suggested two things: that if a person naturally carried a similar mutation in their MSH3 gene, they might also have reduced CAG repeat instability, and hence a better disease prognosis, and secondly, that making drugs to target MSH3 could be valuable for the treatment of Huntington’s disease, assuming that instability is important.
Mismatch repair and the MSH3 protein
So what exactly does MSH3 do, and how might it be capable of impacting the prognosis of Huntington’s disease? To best explain this, we need to learn a little about that ever-so-important biological process mentioned previously, DNA mismatch repair.
DNA repair, in general, is a necessary process that allows our cells to correct mistakes, or mutations, that arise within our genetic code. These mutations can be caused by multiple events, some from normal cellular activities and some from environmental harm, such as UV light or chemicals. Some genetic insults can cause breaks in the DNA, while others introduce single mutations – changes in the sequence of letters in DNA.
Mismatch repair proteins, such as MSH2, MSH3 and MSH6 search out two different types of genetic mistakes: ‘mismatches’ that occur when DNA is being copied in cells that are dividing, and small loops that can form within DNA after a single stand break.
CAG repeats that cause HD are particularly susceptible to the formation of these small loops of DNA. The reason is related to fundamental properties of DNA that make it possible for it to be copied within the cell.
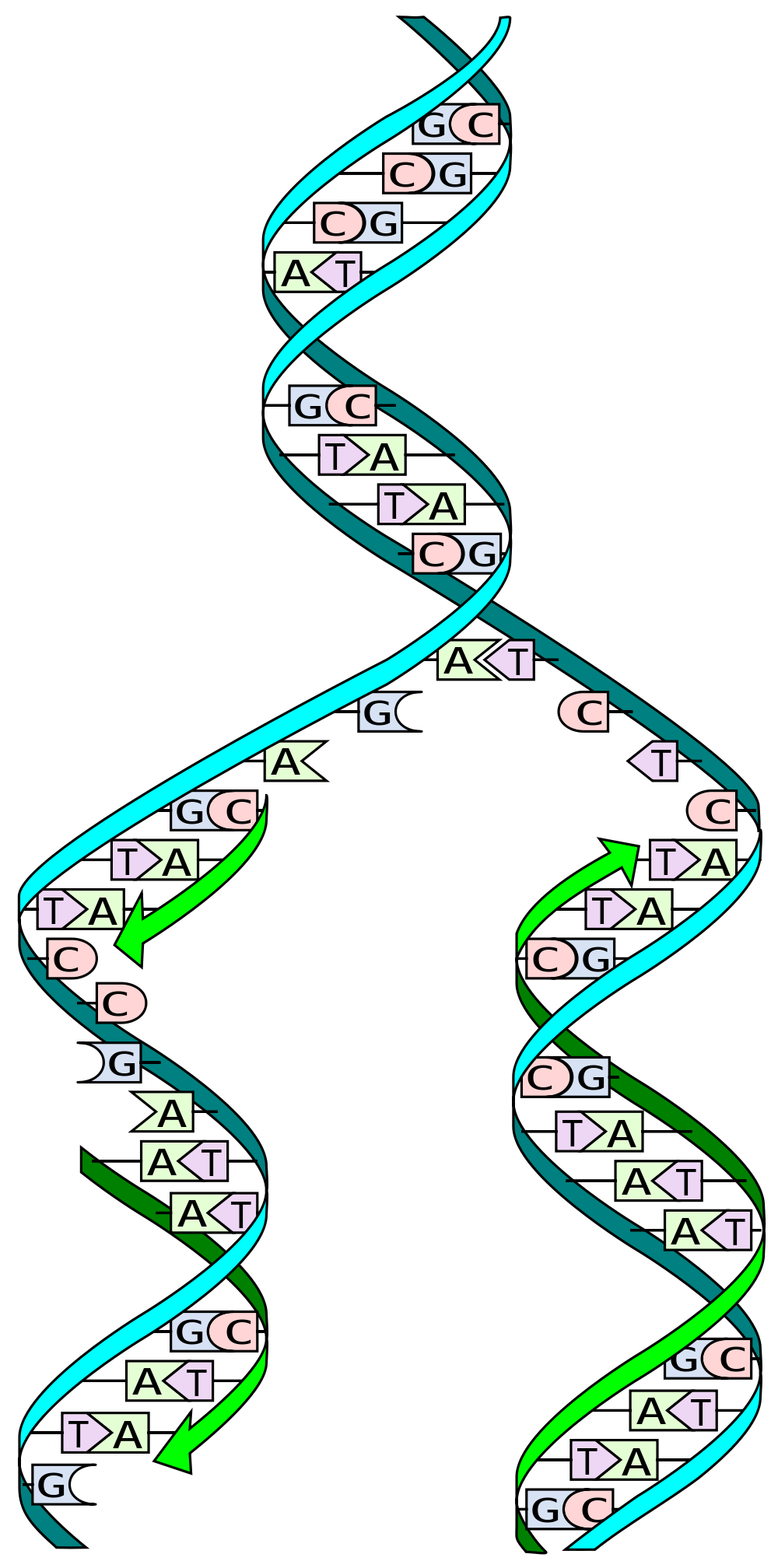
Image credit: Madeleine Price Ball
Many people have heard that DNA is a ‘double helix’. This basically means it made up of two strands that form a coiled structure. These DNA strands are often described as copies of each other, or a mirror image, but this is only partially true.
DNA is made up of only 4 different genetic building blocks called nucleotides, which are named cytosine ©, guanine (G), adenosine (A) and thymidine (T). C has a natural affinity for G, while A is attracted to T.
When DNA is replicated, the double helix is pulled apart into two individual strands. Specialized molecular machines, called ‘polymerases’ then read the genetic code, one letter at a time, generating a new second strand and, in turn, a new copy of the DNA.
To do this, the polymerases exploit the natural affinities of the nucleotides. For example, when the machinery reaches a C in the genetic sequence, it recruits a G into the growing DNA strand, and when it detects a T, it matches it with an A. This is why we say that the two DNA strands aren’t exactly a copy of each other. They provide the information to make the second strand, but they aren’t a replica.
Now, let’s get back to the CAG repeats. When one strand of DNA breaks near or within a long CAG repeat, it can free that srtand to form a loop due to the natural affinity of C’s and G’s within its sequence.
Two mismatch repair proteins, MSH2 and MSH3 come together to search out and repair these types of DNA loops. Unfortunately, one by-product of the repair process is that additional CAG repeats can be erroneously introduced to the sequence. Like a landslide, the more repeats that are added, the greater the original problem becomes. This is the root of genetic instability.
So what exactly is happening in the CBy mice that makes them resistant to repeat expansion? Remember, the researchers found that the CBy mice had a mutation in the genetic sequence of MSH3 that caused the protein to become unstable. With less MSH3 available to find and correct DNA loops within the CAG repeat of the HD gene – possibly introducing extra CAGs in the process – genetic instability ground to a halt.
What’s next?
What does all this mean for Huntington’s disease patients? Right now, all it suggests is that one of the reasons why there is variation in age-of-onset between people with equal repeat lengths could be because of varying ability of their MSH3 proteins to fix these DNA loops. If there were people carrying a similar mutation to the one identified in the mice, they might have delayed disease progression.
In theory, the study also suggests that if we could modify the activity of the MSH3 protein in HD patients, we could modify the amount of repeat instability in their HD gene. If repeat instability is important for the development of HD, this could theoretically slow down the development of the disease.
The challenge of targeting MSH3, however, is that DNA repair is an important process for all the cells in the body. If mutations in genes cannot be corrected efficiently, they have the potential to accumulate and cause cancer. It remains to be seen if disruption of MSH3 activity could be tolerated enough to prevent HD onset, without causing other serious diseases.
This study makes no promises about the therapeutic potential of the research, but they have certainly created interest in tracking MSH3 in patients. The more we know about different genes that can affect Huntington’s disease, the better the information can be used to provide improved patient care through more accurate disease prognosis.
Learn more
For more information about our disclosure policy see our FAQ…