
New work from researchers in London uses mice to narrow in on the number of CAG repeats needed to cause symptoms of Huntington’s disease. Their work points to fewer than 185 CAGs as a threshold.

Replacing cells with HD in the brain could be an effective treatment strategy. Recent work shows that glia injected into mouse brains take over and oust the older cells, but for a surprising reason - because of age, not HD!

Researchers look at the cause and effect of various forms of the HTT protein. They find both expanded and unexpanded HTT contribute to brain cell communication and the brain has an amazing capacity to compensate for changes related to disease

A genetically-tweaked Huntington's disease mouse model shows a tendency for the CAG repeat to grow, just like we see in humans with the mutation.

Researchers with PTC Therapeutics recently published exciting new findings - a promising new huntingtin lowering drug that can be taken as a pill. Will this change how we move forward with huntingtin lowering?

A Chinese research team developed a new way to lower huntingtin protein indirectly, by targeting a protein called GPR52. The molecules they designed were protective in cells and in mice with HD.

Scientists screen the ENTIRE genome to find new potential therapeutic targets for HD. This ambitious study provides a wealth of data for HD researchers

Researchers got surprisingly lucky when looking for drug molecules to pull mutant huntingtin protein into a cellular garbage disposal machine
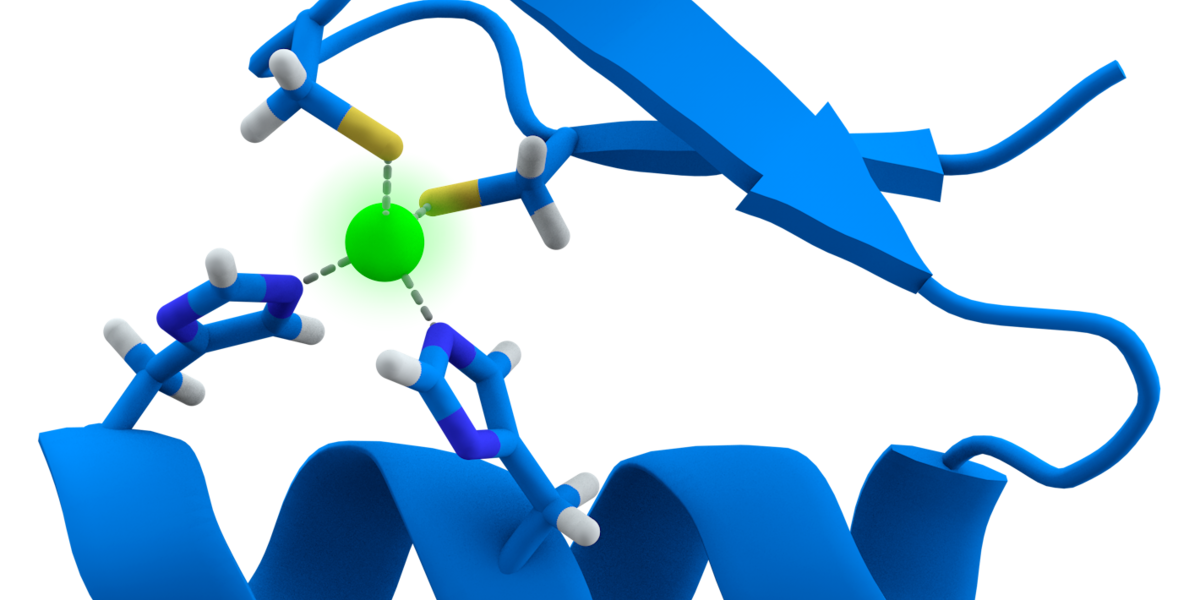
Exciting new Huntingtin Lowering work from @SangamoTx and @CHDIfoundation using "Zinc Fingers" to shut down expression of the mutant Huntingtin gene. More details on this exciting new technique here.