-

Cutting to the chase with CRISPR
Latest News
-

Artificial Intelligence enters the HD space as a diagnostic tool
⏱️9 min read | From predicting symptom onset to tracking movement changes via smartwatch, artificial intelligence tools are being used in research. Here’s where we are, and why Huntington’s disease is a strong candidate for these approaches.
-

Blocking a toxic fragment: new mouse study highlights the importance of HTT1a in Huntington’s disease
⏱️ 9 min read |New research in a HD mouse model points to a key culprit: a small fragment called HTT1a. Lowering HTT1a levels successfully delayed disease signs in mice that model HD, perhaps shaping the next generation of HTT-lowering therapies.
-

Between Care, Genes, and Systems: Navigating Huntington’s Disease as a Caregiver
A small study based in Ireland asked a simple question: what does caring for someone with Huntington’s disease feel like? The answers point to isolation, stigma, and major gaps in healthcare support for many of the respondents.
-

Your Wrist on Watch: Could a Smartwatch Reveal Huntington’s Disease Symptoms?
⏱️ 7 min read | A wrist sensor tracked arm movements in people with HD for a week and could see who had HD and who didn’t. This kind of technology could change how we measure drug effects in trials.
-

March 2026: This Month in Huntington’s Disease Research
⏱️ 9 min read | March brought us the annual CHDI therapeutics conference along with updates from 3 ongoing clinical trials. We also covered new research on somatic expansion and honored the community that built our science during Gratitude Day.
-

Two Heads Are Better Than One: Combined Physical and Music Therapy for Late-Stage Huntington’s Disease
A new study looking into combined music and physical therapy shows that simple rhythmic cues work better than complex music or instructions, helping to improve movement control and reducing chorea.
-

A Guest Perspective for Gratitude Day: Why Huntington’s Disease May Be Neuroscience’s Best Investment
⏱️ 6 min read | Today, on Gratitude Day, we share a guest piece from HD researcher Roy Maimon that Huntington’s disease is neuroscience’s best investment, not just for its scientific clarity, but for the remarkable community built around it.
-
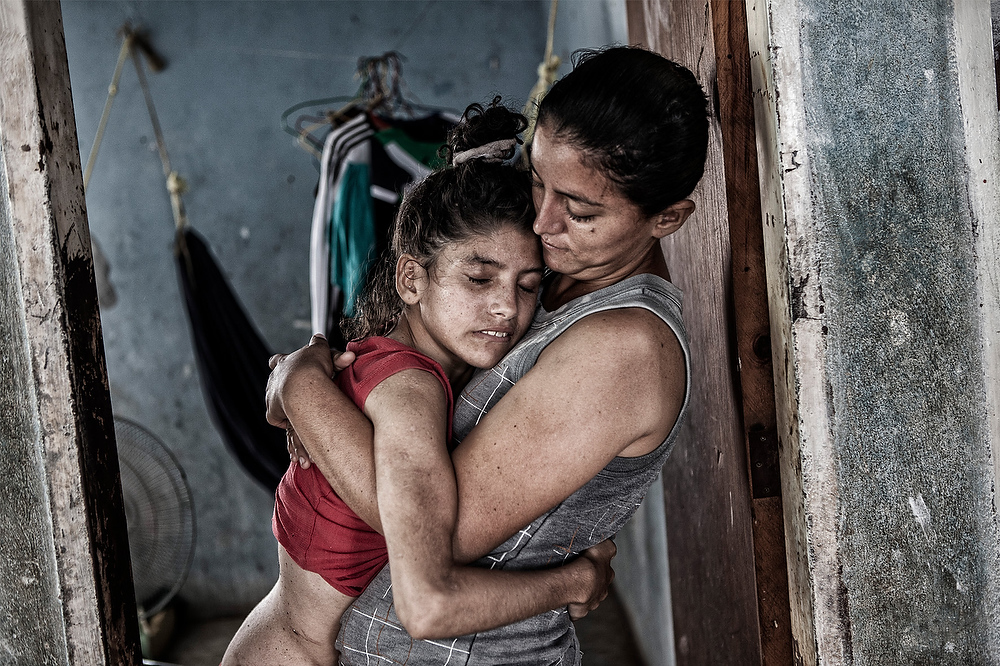
The Girl at the End of the World: A Gratitude Day Reflection on the Place that Built Our Science
⏱️ 9 min read | This Gratitude Day, March 23rd, we celebrate the extraordinary partnership between HD families, scientists, and caregivers, and reflect on the place that built the science on which we all stand.
-

Turning down mismatch repair genes slows Huntington’s repeat growth in human neurons
⏱️ 10 min read | In a human cellular system, scientists dialed back mismatch repair genes to reduce CAG expansion by up to 69%. This work is in its early days, but it shows promise for “anti-expansion” therapies that could be used to delay HD onset.
-

Huntington’s Disease Therapeutics Conference 2026 – Day 3
⏱️ 29 min read | Day 3 of the HD Therapeutics Conference dives into breakthrough science, new tools and bold ideas that are pushing HD research toward better treatments.
HDBuzz Trial Tracker
Sites for Phase 3 trial for votoplam, INVEST-HD, now open. Updates on recruitment and first dosing imminent.
Progress update for NCT06585449 – A Study to Evaluate ALN-HTT02
